頸痛康膠囊的工藝研究
2010-09-11 02:13:56葉玉華劉燦輝
中國藥業 2010年5期
關鍵詞:工藝
葉玉華,劉燦輝
(湖南省邵陽市藥品檢驗所,湖南 邵陽 422001)
頸痛康膠囊的工藝研究
葉玉華,劉燦輝
(湖南省邵陽市藥品檢驗所,湖南 邵陽 422001)
目的 優選頸痛康膠囊的最佳提取工藝。方法 采用正交試驗法,以提取液中葛根素含量及干膏量為指標,綜合評分以優選藥材的提取工藝,并考察成型顆粒的流動性、堆密度、吸濕性及臨界相對濕度。結果 藥材較優的提取工藝為加水煎煮2次,第1次加10倍量水煎3 h,第2次加8倍量水煎2 h;成型顆粒的流動性好,堆密度為0.408 g/cm3,不容易吸濕,臨界相對濕度約為56%。結論 優選的工藝操作簡便,各活性部位提取率高,適用于生產;制得的顆粒間摩擦力小,流動性好,不容易吸濕,可以滿足生產、貯存等需求。
頸痛康膠囊;葛根素;高效液相色譜法;正交試驗
頸痛康膠囊是邵陽市正骨醫院的醫院制劑,由葛根、延胡索、骨碎補、紅花、片姜黃、甘草等藥材組方而成,具有祛風濕、益氣血、補肝腎、化瘀滯的功效,主治頸椎病證屬氣血虧虛、肝腎不足、脈絡閉阻者,可緩解頸項疼痛、沉重、活動不利、頭痛、頭暈、肢端麻木、酸軟乏力等癥狀。筆者根據處方中有效成分的理化性質和藥理作用,以葛根素含量和干膏量為指標,采用正交試驗法,優選藥材的提取工藝條件,并考察了成型顆粒的流動性、堆密度、吸濕性及臨界相對濕度,為生產提供依據。
1 儀器與試藥
島津LC-20A型高效液相色譜儀;AB204-N型電子天平、AG 135型電子天平(梅特勒-托利多儀器<上海>有限公司)。葛根、延胡索、骨碎補等處方中藥材(邵陽市松齡堂中藥飲片有限公司);葛根素對照品(含量測定用,中國藥品生物制品檢定所提供,批號為110752-200410)。甲醇(SCRC國藥集團化學試劑有限公司,批號為20080602)為色譜純,水為重蒸餾水,其余試劑均為分析純。
2 方法與結果
2.1 總固體測定
取處方中除延胡索、紅花、骨碎補外的各味藥材,按1/10處方量稱取各藥材粗片,共9份,按因素水平表加熱煎煮,濾過,濾液濃縮成清膏,定容至200 mL。精密吸取上述清膏液25 mL,置已干燥至恒重的蒸發皿中,水浴上蒸干后,于105℃干燥3 h,置干燥器中冷卻30 min,迅速精密稱定質量。
2.2 葛根素含量測定
2.2.1 色譜條件[1]與系統適用性試驗
色譜柱:UltimateTMXB-C18柱(250 mm ×4.6 mm,5!m);流動相:甲醇 -水(25 ∶75);檢測波長:250 nm;柱溫:35 ℃;流速:1.0 mL/min。在此色譜條件下,葛根素峰與其他組分色譜可達基線分離,且與相鄰色譜峰分離度大于1.5;理論塔板數按葛根素峰計均在4 000以上。
2.2.2 溶液制備
取葛根素對照品適量,精密稱定,加甲醇制成每1 mL含50!g葛根素的溶液,即得對照品溶液。精密吸取上述清膏3 mL,置50 mL量瓶中,加30%乙醇稀釋至刻度,搖勻,濾過,取續濾液,即得供試品溶液。取除葛根外的其余處方量藥材,按確定的制備工藝制備缺葛根的陰性樣品,按供試品溶液制備方法制備缺葛根的陰性樣品溶液。
2.2.3 方法學考察
干擾試驗:分別精密吸取對照品溶液、陰性樣品溶液與供試品溶液各10!L,注入液相色譜儀,測定并記錄色譜圖。結果色譜行為良好,各色譜峰均達基線分離,供試品溶液色譜中在與對照品溶液色譜峰相應位置上檢出色譜峰,而缺葛根的陰性樣品溶液無相應色譜峰,表明陰性樣品對測定無干擾(圖1)。

圖1 高效液相色譜圖
線性關系考察:分別精密吸取質量濃度為0.092 32 g/L的葛根素對照品溶液2,5,10,15,20!L注入液相色譜儀,按上述色譜條件測定峰面積,以對照品質量濃度為橫坐標、峰面積為縱坐標繪制標準曲線,得回歸方程 A=4 777 351.282C+3 830.268,r=0.999 97(n=5)。結果表明葛根素進樣量在0.184 64~1.846 4!g范圍內與峰面積呈良好的線性關系。
加樣回收試驗:精密吸取已知含量的供試品溶液1.5 mL,加入葛根素對照品適量,測定峰面積,計算得平均加樣回收率為100.7%,RSD 為 1.1%(n=9)。
2.3 煎煮工藝研究
2.3.1 正交試驗設計
以加水量(因素A)、煎煮時間(因素B)、煎煮次數(因素C)為考察因素,所得干膏量和葛根素含量為考察指標,按L9(34)表設計正交試驗。因素水平見表1。
2.3.2 試驗方法與結果
測定所得干膏量和葛根素含量,結果見表2。
2.3.3 方差分析
以D項作為誤差項進行方差分析,結果見表3和表4。
2.3.4 結果分析
直觀分析表明,對頸痛康膠囊水煎煮的影響因素為C>A>B。干膏量方差分析結果表明,因素C(煎煮次數)對頸痛康膠囊的水煎煮有非常顯著性影響,因素A(加水量)有顯著性影響;葛根素含量方差分析結果表明,因素C(煎煮次數)對頸痛康膠囊的水煎煮有非常顯著性影響。綜合考慮能源、生產日期等,確定其最佳工藝組合為A3B3(B2)C2,即加藥材8倍量的水浸泡1 h,加熱煎煮2次,第1次 3 h,第 2 次 2 h。
2.4 煎煮液濃縮條件確定
煎煮液應采用減壓濃縮,真空度為-0.07~-0.08 MPa,溫度約80℃,并濃縮至干膏。
2.5 制粒條件確定
經試驗,上述粉末加80%乙醇制得的軟材軟硬適中,顆粒均勻。若加70%的乙醇,則所制軟材太黏,粘附篩網,顆粒太粗;若加90%的乙醇,則所制軟材較松散,難以手握成團,且顆粒粉末太多。

表1 正交試驗因素水平表
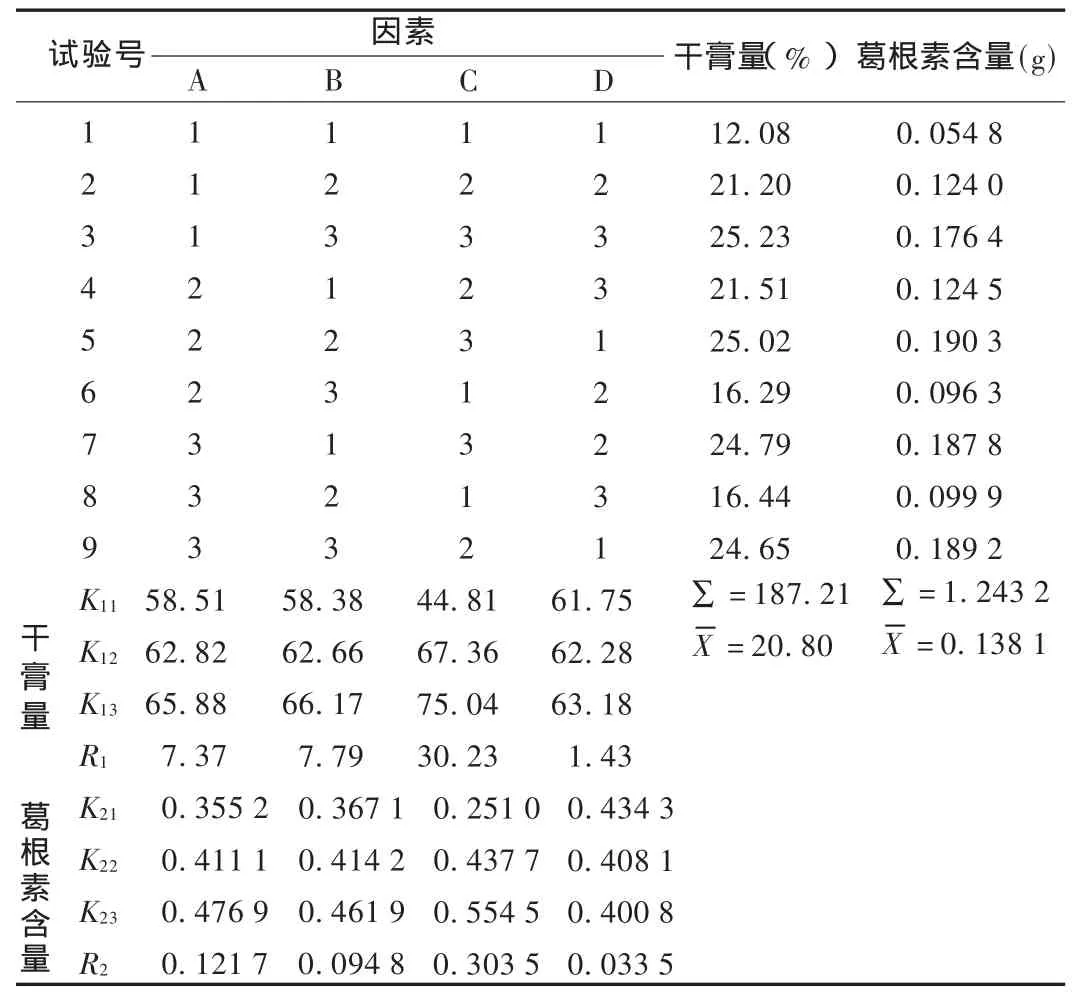
表2 正交試驗結果

表3 干膏量方差分析表

表4 葛根素方差分析表
2.6 顆粒流動性測定[2]
分別取顆粒約50 g,置具1.25 cm小孔的漏斗中,距平板玻璃平面10 cm高度落下,分別測定堆積高度(H)和堆積半徑(r),并按公式tanφ=H/r求出休止角 φ,以 φ表示內容物的流動性,φ愈小,表明流動性愈好。共測3批顆粒,結果批號為070901,070902,070903 的 3 批顆粒休止角分別為 30.28°,30.26°,30.14°,平均30.23°(n=3)。
2.7 顆粒堆密度測定
取顆粒約100 g,通過一個玻璃漏斗緩緩傾倒至一量筒內,測出內容物的松容積(V),并按公式 d=m/V計算出內容物的堆密度 d。共測3批顆粒,結果批號為070901,070902,070903的3批顆粒堆密度分別為 0.410,0.417,0.398 g/cm3,平均 0.408 g/cm3(n=3)。
2.8 顆粒吸濕百分率測定[3]
取底部盛有氯化鈉過飽和溶液的玻璃干燥器,放入25℃的恒溫培養箱內恒溫24 h(相對濕度75%)。在已恒重的稱量瓶底部放入厚約2 mm的顆粒,準確稱重后置盛有氯化鈉過飽和溶液的玻璃干燥器內(稱量瓶蓋打開),25 ℃恒溫培養箱保存,于 2,4,8,12,24,48 h后分別稱量,計算吸濕百分率。吸濕百分率=(吸濕后顆粒質量-吸濕前顆粒質量)/吸濕前顆粒質量×100%。結果保存 2,4,8,12,24,48h后,測得顆粒吸濕百分率分別為3.11%,4.68%,6.86% ,8.45% ,9.87% ,10.57%(圖 2)。
2.9 顆粒臨界相對濕度(RHC)測定[3]
取顆粒干燥至恒重后,置已恒重的稱量瓶中(厚度約2 mm),準確稱量后置于分別盛有表5所列7種不同鹽的過飽和溶液的干燥器內(稱量瓶蓋打開),于25℃恒溫培養箱中保持4 d后稱量,計算吸濕百分率。以吸濕百分率數據為縱坐標、相對濕度數據為橫坐標作圖,得頸痛康顆粒的RHC約為56%。詳見表5和圖3。

圖2 吸濕百分率測定圖
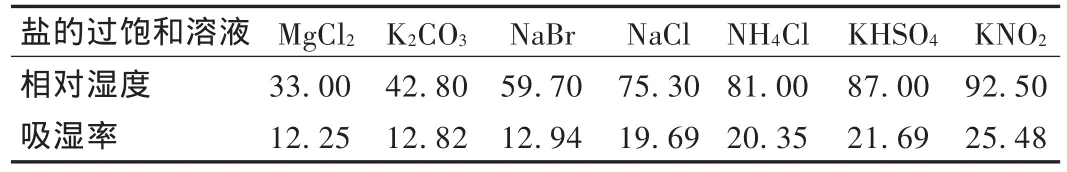
表5 顆粒RHC測定結果(%)
2.10 工藝條件驗證
取10倍處方量的藥材共3份,按優選的工藝條件進行驗證,出膏率分別為17.9%,18.3%,18.0%,RSD=1.3%;葛根素含量分別為1.76,1.64,1.79 mg/粒,RSD=0.82%。結果表明該工藝重現性好,穩定可行。
3 討論
方中葛根為君藥,參照《中國藥典》葛根素含量測定方法,用高效液相色譜法測定該制劑中的葛根素含量,結果方法準確性好,精密度高,故選擇葛根素含量作為正交試驗的一個考察指標。
通過測定顆粒休止角及堆密度,說明顆粒之間的摩擦力較小,流動性較好,可以滿足生產過程中流動性的需求。顆粒的臨界相對濕度約為56%,表明該藥物不容易吸濕,將藥物貯存在其臨界相對濕度以下的環境,能夠延長藥物吸濕平衡到達的時間。
試驗結果表明,該煎煮工藝可行,重現性好,可操作性強。參考文獻:

圖3 顆粒RHC測定圖
[1]國家藥典委員會.中華人民共和國藥典(一部)[M].北京:化學工業出版社,2005:233.
[2]曹春林.中藥藥劑學[M].第5版.上海:上海科學技術出版社,2003:68-71.
[3]劉雪梅,韋文俊,王志萍,等.改善十味扶正顆粒吸濕性的實驗研究[J].中成藥,2006,28(10):1 523-1 524.
TQ461
A
1006-4931(2010)05-0038-02
2009-03-24)
猜你喜歡
中國特種設備安全(2022年5期)2022-08-26 09:19:32
礦產綜合利用(2020年1期)2020-07-24 08:50:40
山東冶金(2019年6期)2020-01-06 07:45:54
收藏界(2019年2期)2019-10-12 08:26:06
世界農藥(2019年2期)2019-07-13 05:55:12
世界農藥(2019年2期)2019-07-13 05:55:10
模具制造(2019年3期)2019-06-06 02:11:00
山東工業技術(2016年15期)2016-12-01 05:30:59
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
