腦顏面血管瘤臨床及影像學 1例報告
2010-09-20 08:03:52鄧麗影
中風與神經疾病雜志 2010年1期
關鍵詞:癲癇
劉 萍, 鄧麗影
腦顏面血管瘤又稱Sturge-Weber綜合征,合并眼部血管異常時又稱為“腦眼顏面血管瘤”,為一種特殊類型的腦血管畸形,系先天性神經皮膚綜合征。本病以顏面血管瘤和癲癇發作為主要臨床特征,可因顱內出血或癲癇持續狀態而威脅生命。
1 病例摘要
患者,女,18歲,因左肢無力 1w于 2006年8月15日就診,患者 8月初右足腫脹,當地診斷“炎癥”,治療好轉,但近1w出現左肢無力,無頭痛及嘔吐,不發熱,無抽搐。既往史:出生時即見右側面部“紅痣”,10歲左右有過抽搐,余無異常。爺奶為表親。體檢:右面三叉神經 1、2支分布區皮膚紅色,微突起(見圖1),皮膚科診斷“海綿狀血管瘤”,左肢肌力3~4級,就診次日未服藥肌力恢復至 5級。
CT示右側半球沿腦回呈曲線形鈣化,雙側腦室脈絡叢增大鈣化(見圖2、圖3),MRI示右側大腦半球較左側小,雙側側腦室三角區可見對稱性球型結節呈 T2高信號,雙顳葉表面見粗大流空信號,左顳骨、頂骨、枕骨斜坡及碟骨板障增厚,T2明顯低信號。T2Flair脈絡叢結節為高信號,DWI為低信號。增強后雙側脈絡叢結節顯著強化,右頂、枕葉腦回狀強化(見圖4、圖5),左側大腦半球、腦干、小腦未見異常強化;MRA示頸內動脈及分支未見異常,椎動脈較細、右側明顯,雙顳血管增粗為靜脈血管(見圖6)。診斷:Sturge-Weber綜合征。
2 討 論
1879年,William Allen Sturge首次描述一位 6歲半孩子患有此類疾病,1922年Parkes Weber報告患者可有腦內鈣化。Sturge-Weber綜合征(斯特奇-韋伯綜合征)為原因尚未闡明的一種先天性綜合征通常為散發病例。主要病理變化是在出生時即已存在的三叉神經區葡萄酒色型的焰色痣(毛細血管性或海綿狀血管瘤)、同側枕、頂、顳葉或額葉軟腦膜的血管瘤(以靜脈性為主)及眼球脈絡膜血管畸形。患側大腦半球發育不良或萎縮,神經節細胞減少、變性、神經膠質增生,并伴有腦皮質鈣化。腦皮質,特別是第二三層,毛細血管可有增厚和鈣化。局部發生層狀壞死、神經細胞脫失、萎縮、膠質細胞增生及鈣鹽沉著。
面部三叉神經分布區內紫紅色面痣,為具有診斷意義的體征。首先出現的神經癥狀通常為面部皮損及對側的局限性癲癇。可有智能減退、對側偏盲和對側肢體輕偏癱、萎縮和肢體生長落后于健側。可有面痣同側的凸眼、青光眼、牛眼或視神經萎縮。身體其他部位也可有葡萄酒色皮痣,伴視網膜、腎、肝等血管瘤,還可伴發隱睪、脊柱裂、脊髓空洞癥等。可因顱內出血或癲癇持續狀態而威脅生命。可根據面部典型分布的特征性皮痣作出診斷。頭部X線平片發現面部血管瘤同側的腦內病理鈣化影,呈雙層線條波浪形、腦回形或樹枝形,乃可確診[1]。 CT表現:(1)鈣化常見,多為單側性,始于枕葉,逐漸向前發展,居腦表淺部位,沿腦回呈曲線形或寬大鋸齒狀鈣化;(2)腦皮質萎縮典型,尤以右枕葉萎縮明顯,亦可累及整個大腦半球,腦溝增寬,但腦室不擴大;(3)患側顱骨增厚,頭部不對稱;(4)軟腦膜血管畸形,增強掃描(CE)可強化。(5)同側脈絡叢增大,且顯著強化。鑒別診斷:蛛網膜下腔出血:腦溝內呈高密度影,但 CT值在80HU以下。其他 CT表現及臨床特點亦與 Sturge-Weber綜合征不同[2]。
癲癇發作的患者可以藥物控制癲癇發作。癲癇發作頑固、不能控制而顱內血管損害相對局限者,可考慮手術切除。
該患者為典型的Sturge-Weber綜合征,右側面部血管瘤嚴格按三叉神經第一、二支分布,顱內靜脈血管異常主要分布右頂、枕、顳葉,與文獻不同的是患者合并雙側脈絡叢增大,且顯著強化。患者雖然顱內病變明顯,但臨床癥狀僅為偶發的癲癇或短期的肢體功能障礙,沒有語言障礙、偏盲,這可能與病變以右側半球頂、枕、顳葉皮層表面為主有關。患者由于皮層及脈絡叢血管明顯異常存在顱內出血的可能,因病變范圍廣手術難度大,只能對癥處理。
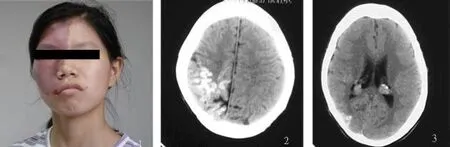
圖1 右側第三叉神經 1、2支分布區酒紅色血管痔; 圖2、圖3 CT:右側半球沿腦回呈曲線形鈣化,雙腦室脈絡叢增大有鈣化;
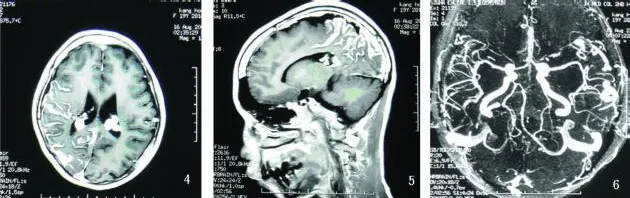
圖4 、圖5 MRI+C右側半球右頂、枕葉腦回狀強化; 圖6 頸內動脈及分支未見異常,椎動脈較細、右側明顯,雙顳血管增粗為靜脈血管
[1]Euen JB,ComiAM,Kossoff EC,etal.Myoclonic-astatic epilepsy in a child with Sturge-Weber syndrome[J].Pediatric Neurology,2007,36(2):115-117.
[2]王文獻,張 冬,劉衛金,等.神經皮膚綜合征的影像學特征和診斷[J].第三軍醫大學學報,2008,30(14):1381-1384.
猜你喜歡
中國民間療法(2021年5期)2021-06-09 09:21:04
中華養生保健(2020年2期)2020-11-16 00:49:00
解放軍醫學院學報(2020年12期)2020-03-29 05:11:46
中成藥(2017年6期)2017-06-13 07:30:35
飲食科學(2017年5期)2017-05-20 17:11:53
臨床醫藥文獻雜志(電子版)(2017年11期)2017-05-17 04:48:10
安徽醫科大學學報(2015年9期)2015-12-16 11:09:44
中國當代醫藥(2015年7期)2015-03-01 02:01:13
西南軍醫(2015年4期)2015-01-23 01:19:30
西部中醫藥(2014年6期)2014-03-11 16:07:47
