氨在肝性腦病發病機制中的作用
2010-11-27 06:20:26張清俊劉雁勇劉赫左萍萍
中國康復理論與實踐 2010年2期
關鍵詞:氧化應激
張清俊,劉雁勇,劉赫,左萍萍
肝性腦病(hepatic encephalopathy,HE)是以臨床上各種嚴重肝臟疾病所致的以代謝紊亂為基礎的中樞神經系統功能失調綜合征。其臨床表現有意識障礙、行為異常、昏迷、撲翼樣震顫、構音障礙、出現病理反射、腦電圖異常、血氨升高等。1998年,第11屆世界消化病協會將肝性腦病重新定義,由不同病因引起的HE分為A、B、C三型,其中將過去稱為亞臨床HE和隱性HE正式命名為輕微 HE(Minimal HE,M HE),將MHE視為HE發病過程中的一個特殊階段[1]。
HE是肝臟解毒功能不全和衰竭表現,其發病機制尚未完全闡明。目前存在的假說主要有:氨中毒、假性神經遞質、GABA、血漿氨基酸失衡、錳中毒等。其中氨中毒學說在HE的發病機制中占主要地位,降血氨是治療HE的有效措施之一。本文結合氨的代謝、特別是病理狀態氨的代謝異常所致氨中毒在HE發病機制中的作用進行綜述。
1 氨在體內的代謝
氨在體內是一種有毒的物質。體內氨的主要來源有:①氨基酸經脫氨基作用產生的氨是主要來源;②由腸道吸收的氨,包括腸內氨基酸在腸道細菌作用下產生的氨和腸道尿素經細菌尿素酶水解產生的氨;③腎小管上皮細胞分泌的氨,主要來自谷氨酰胺酶催化谷氨酰胺的水解。氨在體內的轉運方式有丙氨酸-葡萄糖循環和谷氨酰胺循環。其中,肌肉中的丙酮酸在谷丙轉氨酶的作用下生成丙氨酸將肌肉中生成的氨轉移到肝;腦、肌肉等組織內的氨與谷氨酸在谷氨酰胺合成酶(glutamine synthetase,GS)的作用下生成谷氨酰胺,將氨轉運到肝和腎。氨主要在肝內合成尿素和經腎小管分泌排出體外。
2 氨對機體的影響
2.1 氨引起星形膠質細胞腫脹 星形膠質細胞是大腦中含量最豐富的一種大膠質細胞,主要功能有:①參與血腦屏障結構與功能的完整性;②GS特異性的存在于星形膠質細胞中,參與氨在神經系統的代謝;③支持、營養和保護神經元,引導神經元遷移,分泌神經生長因子等;④細胞膜上有許多神經遞質的轉運體,參與γ氨基丁酸(GABA)、谷氨酸的代謝,還能攝取和滅活單胺遞質,如5-羥色胺、多巴胺、去甲腎上腺素等;⑤調節神經細胞內外離子,維持神經元內外的離子濃度及pH值穩定等。神經膠質細胞酸性纖維蛋白(glial fibrillary acidic protein,GFAP)是星形膠質細胞特有的細胞骨架蛋白,也是星形膠質細胞損傷鑒定的標志。
大量研究表明,氨在體內、外均可引起星形膠質細胞腫脹。臨床檢查結果表明,星形膠質細胞腫脹也是急性肝衰竭時腦水腫的細胞病理基礎。Rama Rao等采用體外神經元細胞和星形膠質細胞共培養技術,觀察到星形膠質細胞可以明顯減輕氨引起的神經元損傷作用[2]。這也提示當星形膠質細胞功能受損時,神經元更容易遭受內外環境改變引起的損傷。Konopacka等研究發現,蛋白激酶PKG在氨引起的星形膠質細胞腫脹過程中起著重要作用,腦內cGMP及血氨的聯合升高會加重腦水腫[3]。Norenberg等對星形膠質細胞的腫脹機理進行總結,包括氧化/硝基化應激、線粒體通透性改變、谷氨酰胺和信號蛋白改變等,認為星形膠質細胞腫脹是多種因素作用的共同結果,其中氨是觸發這些效應的原始誘因[4]。細胞內Ca2+升高、氧化應激、MAP激酶的激活、誘導M PT和激活轉錄因子 NF-κ B,均是由氨作用于神經系統特別是星形膠質細胞而產生的一系列復雜結果,他們對星形膠質細胞功能障礙、HE發病機制和氨神經毒性起著重要作用[5]。
2.2 氨引起的能量代謝障礙 過去的幾十年,人們對氨引起的能量代謝改變進行了大量研究,Rama Rao等[6]總結了氨對機體能量代謝的影響:①糖利用:HE患者腦內葡萄糖的代謝率明顯降低,而在高血氨大鼠動物模型大腦皮層葡萄糖的代謝率亦明顯降低,丘腦和下丘腦葡萄糖的代謝率卻明顯增加。②糖酵解:在高血氨大鼠動物模型大腦糖酵解明顯增加,糖酵解途徑中的酶如磷酸果糖激酶、二磷酸果糖酶、3-磷酸甘油醛脫氫酶和丙酮酸激酶活性增加。在一種先天性高血氨spf小鼠可以持續觀察到腦內葡萄糖含量增加、丙酮酸含量降低,表明其腦內存在糖酵解紊亂。③三羧酸循環:氨消耗三羧酸循環的中間產物α-酮戊二酸、NADH+,合成谷氨酸,導致前兩種物質含量的降低。體外腦線粒體氨處理后,可引起α-酮戊二酸脫氫酶和異檸檬酸脫氫酶活性降低。此外,細胞內蘋果酸-天冬氨酸穿梭系統將NADH從胞漿轉移至線粒體,以確保線粒體內氧化磷酸化的進行。以前認為氨在腦內引起谷氨酸含量的降低是引起蘋果酸-天冬氨酸穿梭系統抑制的誘因,而最近研究表明高氨條件下胞漿和線粒體內天冬氨酸轉氨酶以及線粒體蘋果酸脫氫酶活性降低,對蘋果酸-天冬氨酸穿梭系統也有不利的影響。④線粒體呼吸:對先天性高血氨spf小鼠研究發現,氨可以抑制多種電子傳遞鏈復合體,特別是復合體Ⅳ(細胞色素C氧化酶)。進一步觀察發現,與非突觸內線粒體相比,氨對突觸內線粒體電子傳遞鏈復合體抑制作用更加明顯。⑤ATP的合成及利用:在spf小鼠和氨處理的星形膠質細胞,可以觀察到ATP含量明顯降低。NO能夠激活神經元線粒體和細胞液中多種酶,抑制星形膠質細胞中谷氨酰胺合成酶的活性。Ca2+-CM激活蛋白磷酸酶,抑制蛋白激酶PKC磷酸化,從而激活ATP酶。NMDA受體中過量的Na+被清除,而這一過程消耗大量ATP。細胞液中過量的Ca2+進入線粒體,導致線粒體內Ca2+蓄積,抑制線粒體呼吸,減少ATP合成,增加氧化應激自由基的形成。以上過程導致ATP下降[7]。⑥糖原:糖原是腦內一種重要的能量儲備,在神經活動增加時被分解利用。星形膠質細胞含有大量的糖原。氨處理星形膠質細胞1 d可以短暫引起細胞內糖原含量降低,3 d后可以抑制去甲腎上腺素誘導的糖原分解。這些結果提示,氨抑制細胞內糖原分解導致神經活動增加時能量供應的降低。
2.3 氨引起的線粒體通透性轉換 線粒體通透性轉換(mitochondrial permeability transition,MPT)可能參與氨引起的線粒體失能。MPT是指線粒體內膜對小分子(<1.5 kDa)物質的通透性突然增加,這是由位于線粒體內膜的通透性轉換孔(permeability transition pore,PTP)突然開放引起的。MPT作用的直接結果是使線粒體內膜電位(△Ψm)的消失,△Ψm的消失會引起線粒體基質腫脹、氧化磷酸化不全、ATP合成障礙以及產生活性氧(ROS)。MPT產生的后果可能是細胞的凋亡或壞死。
目前尚未完全闡明PTP的精確構成,已知結構如圖1所示。其結構包括腺苷酸轉位體(ANT)、電壓依賴性陰離子通道(VDAC)、外周型苯二氮 受體(Peripheral-type benzodiazepine receptor,PBR)、癌蛋白Bcl-2、己糖激酶(HK)和肌酸激酶(CK)等。誘發 MPT的因素有線粒體內Ca2+濃度增加、膜上巰基和吡啶核苷酸的氧化還原狀態、基質pH增加、△Ψm的去極化、ROS和一氧化氮(NO)增加、PBR增加等。環孢霉素 A(CsA)可競爭性抑制線粒體基質蛋白CyP-D,可以特異性地抑制M PT。
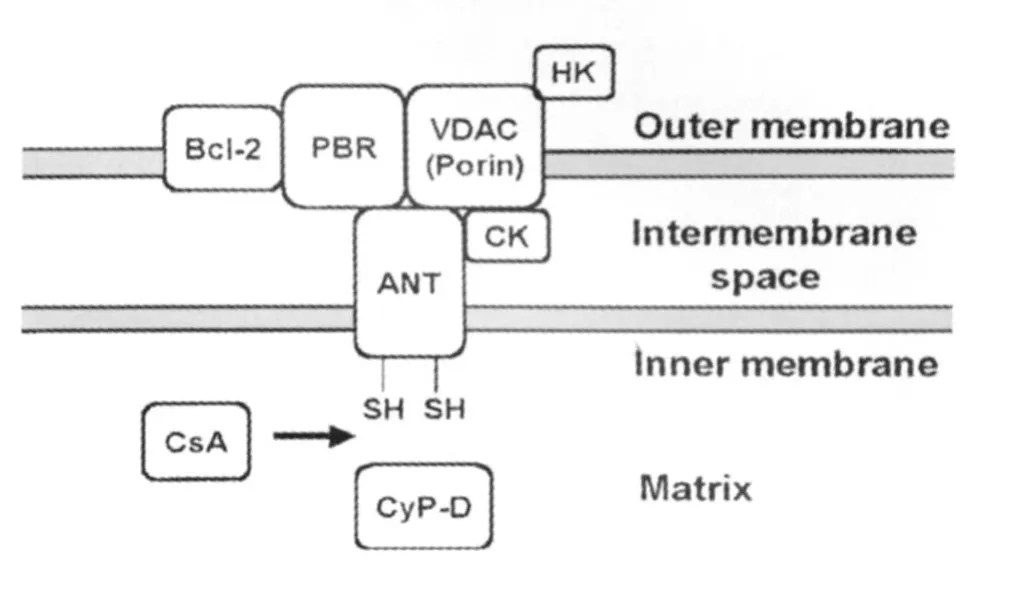
圖1 線粒體通透性轉換孔(PTP)的結構(from Rama Rao,et al.,2003)
Rama Rao等采用多種實驗方法證實氨可以誘導線粒體發生M PT。采用膜電位熒光探針JC-1和TM RE的實驗結果顯示,氨處理培養的星形膠質細胞后引起線粒體△Ψm降低,將細胞與CsA共孵育則可抑制△Ψm降低。采用2-脫氧-6-磷酸葡萄糖(2-DG-6-P)作為指示劑,發現濃度為5 mmol/L的NH4Cl以時間-濃度依賴性方式引起星形膠質細胞的線粒體對2-DG-6-P通透性增加。采用鈣黃綠熒光素作為熒光探針,氯化鈷作為熒光淬滅劑,由于后者不能透過正常的線粒體膜,所以不能淬滅進入正常線粒體的鈣黃綠熒光素,而只有在發生MPT時,氯化鈷才可進入線粒體淬滅其熒光。使用這種方法,可以直接觀察到NH4Cl處理星形膠質細胞24 h后,線粒體內熒光強度明顯降低[8]。以上的研究結果均證明氨可引起線粒體的MPT。
如圖1所示,PBR是線粒體PTP的一個組成亞基,PBR與存在于中樞GABA受體上的苯二氮 受體在分子結構、藥理學特性、體內分布方面均不相同。在HE患者、試驗性肝衰竭動物模型以及氨處理的星形膠質細胞,均發現PBR密度上調。PBR受體增加一方面可能促進神經類固醇激素的產生,后者進一步上調GABAA受體,產生神經抑制作用;另一方面可能誘發線粒體的M PT。Rama Rao等使用nmol級的PBR配體與星形膠質細胞孵育后,可以觀察到PBR誘發的線粒體MPT[8]。PBR誘發線粒體MPT的機制還不明確,可能與PBR配體引起的細胞氧化應激有關[9]。
2.4 氨引起的氧化應激效應 1990年O'Connor和Costell首先發現高血氨小鼠的肝、腦內脂質過氧化物增加以來,氨引起的氧化應激效應隨后受到人們的重視。臨床尸檢發現,HE患者腦內可見一種特征性的AlzheimerⅡ型星形膠質細胞,胞核大而蒼白,核周圍有脂褐素聚積,在這些脂褐素中有大量的過氧化脂質。而在氨處理的星形膠質細胞和視網膜Müller細胞均可發現這種過量的脂褐素樣顆粒。這說明 HE患者腦內的AlzheimerⅡ型星形膠質細胞與氨引起的氧化應激有關。
關于氨引起體外培養的星形膠質細胞中氧化應激的研究比較多。氨可以引起細胞內GSH含量降低,而GSH通過維持蛋白巰基的還原狀態使蛋白質維持正常的空間構象和活性。進一步研究發現,氨可以抑制星形膠質細胞攝取胱氨酸,而胱氨酸是合成GSH的原料,其含量降低預示著細胞更容易遭受氧化應激。氨可以使星形膠質細胞形態發生改變,如星型形狀增強、胞漿空泡、濃縮及嗜堿性增加,而用抗氧化酶SOD和CAT可以完全抑制細胞形態的這種改變。氨引起自由基產生增加的機制還不明確,一種推測是氨激活了N-甲基-D-天冬氨酸(N-methyl-D-aspartate,NMDA)受體,另一種推測是引起線粒體MPT,造成線粒體失能,氧化磷酸化不完全,從而產生大量的ROS。
氨可引起細胞內谷氨酰胺合成增加。谷氨酰胺參與體內氨的解毒、合成谷胱甘肽及核苷酸等。然而高濃度的谷氨酰胺也可以間接導致自由基的產生。用磷酸化激活谷氨酰胺酶(PAG)的抑制劑可以阻斷6.5 mmol/L谷氨酰胺引起的星形膠質細胞自由基產生,PAG抑制劑可抑制谷氨酰胺分解為氨和谷氨酸,這說明谷氨酰胺在分解時產生的氨可誘發自由基的產生。在高氨環境中的星型細胞內,過多的谷氨酰胺被線粒體內的PAG分解,在線粒體內產生高濃度的氨,造成線粒體產生MPT,進而產生 ROS。谷氨酰胺以“特洛伊木馬”的方式侵入線粒體,釋放出氨,誘導線粒體ROS產生,最終導致谷氨酰胺的神經氧化應激損傷效應。
如前所述,在HE患者、試驗性肝衰竭動物模型以及氨處理的星形膠質細胞,均發現PBR密度上調。進一步研究發現,PBR激動劑可以直接導致星形膠質細胞自由基的產生。在培養的星型細胞和神經元細胞中,分別加入10 nM的PBR激動劑如PK11195、Ro5-4864和PpIX后,結果發現,對于星形膠質細胞,所有的激動劑作用2 min均可導致自由基產生的增加;對于神經元細胞,PK11195和PpIX作用2 min僅導致自由基產生的短暫增加,而Ro5-4864無作用,這種差別可能與 PBR在不同細胞內的分布差異有關[10]。
氨引起氧化應激反應,通過酪氨酸殘基硝基化、星形膠質細胞和神經元細胞 RNA氧化導致蛋白質修飾。氨導致的RNA和蛋白質氧化可能減少突觸后蛋白質合成,影響學習記憶功能。RNA氧化為HE患者神經遞質系統和基因表達的多重紊亂以及認知障礙提供了新的解釋[11]。
2.5 氨引起GABA能神經遞質的改變 GABA是一種抑制性神經遞質。進入線粒體的氨與α-酮戊二酸結合成谷氨酸,然后在谷氨酸脫羧酶的作用下生成GABA,通過激活GABA受體產生生物效應。GABA受體分為GABAA、GABAB和GABAC3種類型。GABAA受體主要分布在腦內,激活后使氯離子內流增加,導致突觸后膜超極化,從而產生神經抑制作用。GABAA受體復合物的主要調控點包括苯二氮 (BZ)、巴比妥類、神經類固醇和印防己毒素等。
氨可以直接與GABAA受體復合物相互作用,增強GABA能神經遞質的作用。利用放射配體結合試驗,發現0.05~0.5 mmol/L的氨可提高蠅蕈醇(GABAA受體激動劑)和氟硝西泮(苯二氮 受體激動劑)與GABAA/BZ受體的親和力。細胞試驗表明,0.2~0.5 mmol/L的氨可劑量依賴性地誘發GABA(10-5M)介導的神經元Cl-內流,其原因可能是氨參與增強 GABA與GABAA受體的親和力[12]。
用NH4Cl處理星形膠質細胞,可抑制GABA的攝取,同時增加GABA的釋放[13]。氨的這種效應使細胞外的GABA濃度增加,從而激活GABAA受體。此外,如前所述,氨可以使PBR上調,PBR水平升高能促進神經類固醇激素的產生,而神經類固醇激素能上調GABAA受體密度,產生神經抑制作用。
2.6 氨引起Glu能系統神經的改變 與GABA神經遞質相反,Glu是中樞神經系統的興奮性神經遞質。Glu受體分為兩種主要類型:離子型谷氨酸受體(iGluR)和代謝型谷氨酸受體(mGluR)。iG1uR根據受體選擇性激動劑的不同,可進一步分為 N-甲基-D-天冬氨酸(NM DA)受體、α-氨基-3-羥基-5-甲基-4-異 惡 唑 (α-amino-3-hydroxy-5-methyl-4-isoxazolepopionate,AMPA)受體和海人藻酸(kainate,KA)受體。
Monfort等總結高氨對Glu神經遞質及其受體的影響:①氨可以引起Glu的釋放和攝取改變,這種改變的結果導致細胞外間隙Glu濃度的增加,最終導致Glu受體的激活;②氨可以引起星形膠質細胞膜的谷氨酸轉運體(GLT)表達降低;③高濃度的氨可以直接激活NMDA受體。NMDA受體激活后導致Ca2+的內流,Ca2+與鈣調素結合,激活一氧化氮合酶(NOS),導致NO合成增多,NO可以激活鳥苷酸環化酶,最終導致細胞內cGM P濃度增加,引起進一步的細胞效應;④慢性長期中濃度的氨可以引起NMDA受體結合位點的降低,這可能是氨引起細胞間隙Glu濃度增高的代償反應;⑤不同濃度和作用時間的氨對NMDA功能的直接作用不同。1 mmol/L的氨可以通過抑制蛋白激酶C(PKC)磷酸化,抑制NMDA受體的激活,而0.1 mmol/L的氨沒有這種作用[14]。
2.7 氨引起的細胞信號傳導通路的改變 絲裂原活化蛋白激酶(mitogen-activated protein kinases,MAPKs)與細胞膜表面的生長因子受體耦聯,也可被細胞應激激活。M APK家族通過磷酸化作用激活酶、轉錄因子、結構蛋白以及其他細胞內信號因子,其成員包括ERK,p38-MAPK和JNK等。Jayakumar等觀察到,用NH4Cl處理星形膠質細胞,可以激活MAPK通路,用抗氧化劑可以阻止這種激活效應,并且 MAPK通路阻滯劑可以使星形膠質腫脹的程度減輕[15]。
核轉錄因子(nuclear factor-kappa B,NF-κ B)是體內廣泛存在的一種即早刻轉錄因子,通過激活細胞因子、細胞黏附分子、生長因子和免疫受體等的表達發揮作用。NF-κ B通常由P65和P50亞基以異源二聚體的方式與抑制分子I-κ Bα結合,存在于胞漿中。 當 I-κ Bα被磷酸化后,I-κ Bα與 NF-κ B分離,NF-κ B轉位進入細胞核,刺激多種基因的轉錄。Sinke等發現5 mmol/L的 NH4Cl處理星形膠質細胞可以引起 NF-κ B轉位的增加,而采用抗氧化酶、維生素E以及p38-MAPK的特異性抑制劑均可以降低 NF-κ B的轉位,表明氧化應激、M APK信號通路參與氨引起的 NF-κ B激活[16]。NF-κ B的激活還可以促進誘導型一氧化氮合酶(iNOS)表達的增加[17],進而促進一氧化氮(NO)的合成,后者可以導致星形膠質細胞腫脹。采用NF-κ B抑制劑BAY11-7082和 SN-50可以抑制氨引起的NO產生及細胞腫脹。
此外,氨還可以直接引起細胞內Ca2+濃度增加[18],激活細胞內Ca2+依賴性蛋白酶(如 NOS)產生自由基,促進 Glu釋放等。
2.8 氨對水通道蛋白4表達的作用 水通道蛋白4(Aquaporin 4,AQP4)屬于特異性的水通道蛋白家族成員之一。人類AQP4基因定位于染色體18q11.2和q12.1連接處,編碼大約31 kDa和34 kDa兩種蛋白異構體,兩者在大腦均有表達,但前者主要存在于其他組織。AQP4在星形膠質細胞含量豐富,特別是在星形膠質細胞與皮質毛細血管相對應的終足部位。這種巧妙的定位說明AQP4對血腦屏障水的通透性調節具有特殊的作用。
Rama Rao等研究發現,用 5 mmol/L的 NH4Cl處理星形膠質細胞,10 h后就可觀察到AQP4表達水平增加,并持續到48 h,細胞腫脹出現在給藥12 h后[19]。相關分析結果表明,AQP4表達的增加與細胞腫脹之間存在相關性(r2=0.95)。進一步研究發現,CsA可劑量依賴性地抑制氨引起的AQP4表達增加,同時也能抑制氨引起的膠質細胞腫脹,說明線粒體MPT可能參與氨引起的AQP4表達增加[20]。此外,氧化應激、錳均參與AQP4表達調節[21]。
3 小結
氨可以通過引起能量代謝、氧化應激、線粒體MPT、GABA和Glu神經遞質系統、細胞內信號轉導以及 AQP4表達的改變,導致星形膠質細胞水腫,使星形膠質細胞對神經元的營養支持作用降低,這些因素綜合作用的結果是產生HE。降氨、促進氨排泄治療已是臨床治療HE的有效手段,明確氨在HE發病機制的作用,可為進一步開發抗氨毒性的HE治療藥物提供新的思路。
[1]Ferenci P,Lockwood A,Mullen K,et al.Hepatic encephalopathy-Definition,nomenclature,diagnosis,and quantification.Final Report of the Working Party at the 11th world Congresses of Gastroenterology,Vienna,1998[J].Hepatology,2002,35(3):716-721.
[2]Rama Rao KV,Panickar KS,Jayakumar AR,et al.Astrocytes protect neurons from ammonia toxicity[J].Neurochem Res,2005,30(10):1311-1318.
[3]Konopacka A,Konopacki FA,Albrecht J.Protein kinase G is involved in ammonia-induced swelling of astrocytes[J].J Neurochem,2009,109(Suppl 1):246-251.
[4]Norenberg MD,Jayakumar AD,Rama Rao KV,et al.New concepts in the mechanism of ammonia-induced astrocyte swelling[J].Metab Brain Dis,2007,22:219-234.
[5]Norenberg M D,Rama RKV,Jayakumar A R.Signaling factors in the mechanism of ammonia neurotoxicity[J].Metab Brain Dis,2009,24(1):103-117.
[6]Rama Rao KV,Norenberg MD.Cerebral Energy Metabolism in Hepatic encephalopathy and Hyperammonemia[J].Metab Brain Dis,2001,16(1/2):67-78.
[7]Rodrigo R,Cauli O,Boix J,et al.Role of NMDA receptors in acute liver failure and ammonia toxicity:therapeutical implications[J].Neurochem Int,2009,55(1-3):113-118.
[8]Rama Rao KV,Jayakumar A R,Norenberg M D.Ammonia eurotoxicity:Role of the Mitochondrial Permeability Transition[J].Metab Brain Dis,2003,18(2):113-176.
[9]Jayakumar AR,Panickar KS,Norenberg MD.Effects on free radical generation by ligands of the peripheral benzodiazepine receptor in cultured neural cells[J].J Neurochem,2002,83:1226-1243.
[10]Norenberg MD,Jayakumar AR,Rama Rao KV.Oxidative Stress in the Pathogenesis of Hepatic Encephalopathy[J].Metab Brain Dis,2004,19(3-4):313-329.
[11]Haussinger D,Gorg B.Interaction of oxidative stress,astrocyte swelling and cerebral ammonia toxicity[J].Curr Opin Clin Nutr Metab Care,2010,13(1):87-92.
[12]Jones EA.Ammonia,the GABA neurotransmitter system,and hepatic encephalopathy[J].Metab Brain Dis,2002,17(4):275-281.
[13]Bender AS,Norenberg M D.Effect of ammonia on GABA uptake and release in cultures astrocytes[J].Neurochem Int,2000,36:389-395.
[14]Monfort P,Munoz MD,ElAyadi A,et al.Effects of hyperammonemia and liver failure on glutamatergic neurotransmission[J].Metab Brain Dis,2002,17(4):237-250.
[15]Jayakumar AR,Panickar KS,Murthy ChRK,et al.Oxidative stress and MAPK phosphorylation mediate ammoniainduced cell swelling and glutamate uptake inhibition in cultured astrocytes[J].J Neurosci,2006,26:4774-4784.
[16]Sinke AP,Jayakumar AR,Panickar KS,et al.NFκ B in the mechanism of ammonia-induced astrocyte swelling in culture[J].J Neurochem,2008,106:2302-2311.
[17]Kleinert H,Pautz A,Linker K,et al.Regulation of the expression of inducible nitric oxide synthase[J].Eur J Pharmacol,2004,500:255-266.
[18]Rose C,Kresse W,Kettenmann H.Acute insult of ammonia leads to calcium-dependent glutamate release from cultured astrocytes,an effect of pH[J].J Biol Chem,2005,280:20937-20944.
[19]Rama Rao KV,Chen M,Simard JM,et al.Increased aquaporin-4 expression in ammonia-treated cultured astrocytes[J].Neuroreport,2003,14:2379-2382.
[20]Rama Rao KV,Chen M,Simard JM,et al.Suppression of ammonia-induced astrocyte swelling by cyclosporin A[J].J Neurosci Res,2003,74:891-897.
[21]Rama Rao KV,Norenberg MD.Aquaporin-4 in hepatic encephalopathy[J].Metab Brain Dis,2007,22:265-275.
猜你喜歡
中成藥(2021年5期)2021-07-21 08:39:04
世界科學技術-中醫藥現代化(2020年2期)2020-07-25 02:05:56
中成藥(2018年6期)2018-07-11 03:01:24
中成藥(2018年5期)2018-06-06 03:11:43
天然產物研究與開發(2016年6期)2016-06-05 10:29:26
西南軍醫(2016年6期)2016-01-23 02:21:19
新疆醫科大學學報(2015年10期)2015-12-26 12:33:30
吉林大學學報(醫學版)(2015年4期)2015-12-17 07:48:13
實用中西醫結合臨床(2015年7期)2015-02-28 16:30:23
癌變·畸變·突變(2015年3期)2015-02-27 06:15:09
