不同濃度醇提取淫羊藿功效成分的研究
2011-01-23 03:38:30嚴躍進金燕敏張蒞峽
中國中醫(yī)基礎醫(yī)學雜志 2011年5期
關鍵詞:黃酮
嚴躍進,沈 勇,黃 偉,金燕敏,張蒞峽
(海南亞洲制藥有限公司藥物研究所,浙江 金 華 321017)
淫羊藿為常用中藥,為小檗科(Berberidaceae)淫羊藿屬(Epimedium)多種植物的干燥地上部分。始載于《神農本草經(jīng)》,別名仙靈脾。《本草綱目》中記載:莖葉入藥,辛溫無毒,具有“益精氣,堅筋骨,補腰腎,強心力”等功效[1]。現(xiàn)代研究證明,淫羊藿苷、淫羊藿總黃酮、淫羊藿多糖是淫羊藿的主要有效成分。此外,尚還有生物堿、甾醇、卅一烷及維生素E等[2、3],其主要有補腎壯陽、祛風濕、抑制微生物、擴張冠脈、抗衰老、促進骨細胞生長以及促進免疫等作用[4~6]。本文針對淫羊藿提取工藝研究中經(jīng)常遇到的問題,采用不同濃度乙醇用冷滲漉法,考察了其提取工藝對淫羊藿苷、淫羊藿總黃酮及淫羊藿總多糖含量的比較研究。
提取淫羊藿中的主要成分——淫羊藿苷、總黃酮類及多糖等,通常采用的方法有乙醇熱回流法、乙醇滲漉法、堿提取法、水提醇沉法等。但是由于在乙醇熱回流法中,需要反復加熱、多次回流,水提醇沉法中因為濃縮時間過長,這些都會使有效成分被破壞,無效雜質成分增多;在堿提取法中,提取和濃縮時,會發(fā)生苷類成分堿水解,使有效成分提取量減少。這些方法都有不足之處。所以我們采用乙醇冷滲漉的方法,選用不同濃度乙醇(20%、50%、70%和95%),采用相同流速滲漉,以3個定量指標進行對比試驗,以選取最佳乙醇提取濃度。這樣可以使苷類成分損失較少,并且有效成分很少會被破壞,應當說這是最理想的提取方法
1 材料
SK3200H型超聲處理器(120W,59HZ)(上海科導超聲儀器有限公司);高效液相色譜儀(HPLC)HP-1100型(Agilwnt Technologies);色譜柱Kromasil C18(250×4.6mm 5mm serial:8511233);紫外分光光度計(島津UV-2550);電子天平FA1004(上海精料天平公司);淫羊藿苷(中國藥品生物制品鑒定所,批號0737-9709);D-無水葡萄糖(中國藥品生物制品鑒定所,批號110833-200302);用于HPLC法測定的試劑均為色譜純,水為重蒸餾水;用于分析的試劑均為分析純,水為蒸餾水;淫羊藿(購于四川綿陽,經(jīng)鑒定為箭葉淫羊藿)。
2 方法
2.1 冷浸-滲漉提取法
經(jīng)過多次預試驗并結合實際情況,我們設計了4個試驗點:分別為20%、50%、70%和95%4個不同濃度乙醇提取液,作為選擇以下相同實驗條件,分段進行提取。
各稱取500g經(jīng)粉碎過篩的淫羊藿樣品,用設計濃度的乙醇潤濕2h,浸泡20h,并分別以上述濃度乙醇作為溶媒,按6.0ml/kg.mim的流速,滲漉至體積為3000ml(每取500ml滲漉液為1份,同濃度下按先后依次編號1#~6#)。分別減壓濃縮直至干燥,得提取物稱重(結果見表1及圖1),測淫羊藿苷(結果見表2及圖2)及淫羊藿總黃酮(結果見表3及圖3)。最后各用3000ml水滲漉,得水滲漉液,減壓濃縮、干燥,得提取物、稱重,測總多糖(結果見表4及圖4)。

表1 4種乙醇濃度提取的提取物重量(g)

圖1 4種濃度乙醇提取的提取物重量
2.2 生藥的含量測定
2.2.1 方法 按照《中國藥典》2005年版淫羊藿項下的檢測方法[7]。
2.2.2 結果 測得淫羊藿苷的含量為0.69%,淫羊藿總黃酮含量是7.11%,表明藥材符合《中國藥典》規(guī)定。
2.3 醇提取中淫羊藿苷、淫羊藿總黃酮以及水提取中總多糖的測定
2.3.1 淫羊藿苷的含量測定(高效液相色譜法)色譜條件:色譜柱:Kromasil C18(250×4.6mm 5mm serial:8511233);流動相:乙腈-水(30∶70);流速:1.0ml/min;柱溫:30℃;檢測波長:UV270nm;進樣量:10μl。
對照品溶液制備及標準曲線:精密稱取淫羊藿苷對照品13.29mg,至25ml容量瓶中,用甲醇溶解并稀釋至刻度,搖勻,再分別精密量取1ml~5ml至10ml容量瓶中,用甲醇稀釋至刻度搖勻。按上述色譜條件進樣,以樣品濃度C為橫坐標,峰面積A為縱坐標。繪制標準曲線,得回歸方程A=81212C-158.36,r=0.9991,表明淫羊藿苷濃度在0.05316-0.2658mg/ml范圍內與峰面積呈良好線性關系。
淫羊藿苷測定用供試溶液的制備及測定:精密稱取樣品約0.2g,置具塞錐形瓶中,精密加入稀乙醇20ml稱定重量,超聲處理1h再稱定重量,用稀乙醇補足減失的重量,搖勻、濾過取續(xù)濾液即得。隨后在上述條件下測定并計算得結果(詳見表2及圖2)。

表2 不同乙醇濃度從淫羊藿中提取淫羊藿苷的重量(g/500ml)

圖2 不同乙醇濃度從淫羊藿中提取淫羊藿苷的重量(g/500ml)
2.3.2 淫羊藿總黃酮的含量測定(紫外分光光度法)紫外條件:島津UV-2550型分光光度計,檢測波長:UV270nm。
對照品溶液制備及標準曲線:精密稱量取淫羊藿苷對照品13.29mg,至25ml容量瓶中,用甲醇溶解并定容至刻度,搖勻。即得對照品溶液。精密量取對照品溶液0.3、0.5ml及1~6ml,分別至50ml容量瓶中,甲醇稀釋至刻度搖勻。以甲醇為空白,270nm波長處測定樣品中淫羊藿總黃酮吸收度。以樣品濃度C為橫坐標,峰面積A為縱坐標。繪制標準曲線,得回歸方程A=40.79379C-0.00135,r=0.9994,表明淫羊藿總黃酮濃度在0.0031896-0.063792mg/ml范圍內與吸收度呈良好線性關系。
淫羊藿總黃酮測定用供試溶液的制備及測定:精密量取淫羊藿苷測定項下供試溶液0.5ml,置50ml容量瓶中,加甲醇至刻度搖勻,作為供試溶液。隨后在上述條件下測定并計算結果(詳見表3及圖3)。
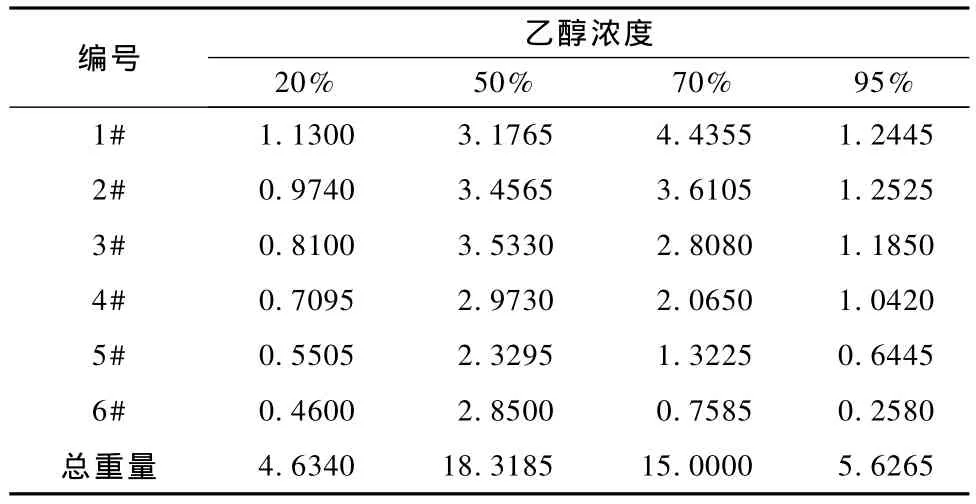
表3 4種不同乙醇濃度的提取物中總黃酮的重量(g/500ml)
2.3.3 總多糖的含量測定(DNS法)紫外條件:島津UV-2550型分光光度計,檢測波長UV520nm。
對照品溶液制備及標準曲線:精密稱量在105℃干燥恒重的無水葡萄糖對照品約50mg,置于50ml容量瓶中,加水溶解并稀釋至刻度搖勻,即得對照品溶液。經(jīng)0.45μm濾膜過濾,棄取初濾液,取續(xù)濾液5ml,置于25ml容量瓶中,加水稀釋至刻度搖勻。以水為空白,520nm波長處測定樣品中淫羊藿總多糖吸收度。以樣品濃度C為橫坐標,峰面積A為縱坐標。繪制標準曲線,得回歸方程A=5.67840C-0.22923,r=0.9992,表明淫羊藿總多糖濃度在0.04659-0.23296mg/ml范圍內與吸收度呈良好線性關系。
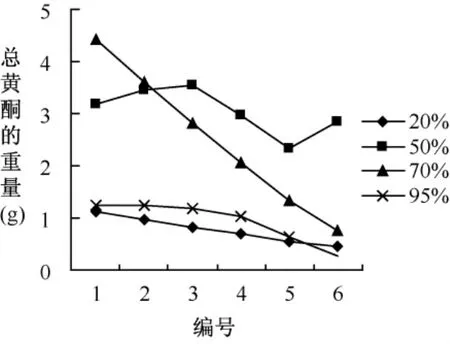
圖3 不同乙醇濃度從淫羊藿中提取淫羊藿總黃酮的重量(g/500ml)
還原糖供試品的制備:精密稱取約35mg,置25ml容量瓶中,加水20ml超聲處理,使完全溶解,定容至刻度,搖勻、濾過,取續(xù)濾液即得。
總多糖供試品的制備:精密稱取約35mg,置50ml容量瓶中,加鹽酸溶液(1->2)10ml,置沸水浴中10min取出,放冷后加40%氫氧化鈉調PH至中性,加水至刻度搖勻、濾過,取續(xù)濾液即得。

表4 不同濃度乙醇處理后水提取物中總多糖含量(%)
總多糖的測定:精密量取上述對照品溶液,還原糖和總糖供試液各2ml。分別置10ml容量瓶中,各加入3,5-二硝基水楊酸試液1.5ml搖勻,置沸水浴中加熱5min取出,立即放入冰水中冷卻30min后,加水至刻度搖勻,在上述條件下測定并計算得結果(見表4及圖4)。
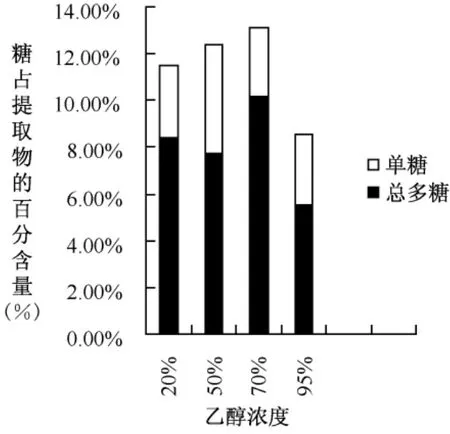
圖4 不同濃度乙醇處理后水提取物中總多糖含量
3 小結
針對不同的提取要求,采用不同的乙醇濃度。經(jīng)過比較分析得出結論:提取淫羊藿苷為主時,宜選擇用70%乙醇濃度提取;提取淫羊藿總黃酮為主,選擇50%乙醇濃度為最佳;在相同得條件下,總多糖得提取則應該選擇用70%濃度乙醇提取后,再用水滲漉得方法。
4 討論
4.1 乙醇提取
圖1顯示:(1)20%和50%乙醇濃度提取時,當取滲漉液為3倍量時,達到最高值;(2)70%乙醇濃度提取時,滲漉液倍量呈線性降低趨勢;(3)50%和95%乙醇濃度滲漉出的醇溶物重量分別為最高和最低。
4.2 從淫羊藿中提取淫羊藿苷
圖2顯示:(1)50%和70%乙醇提取時,隨著滲漉液的不同,倍量呈線性降低趨勢;(2)20%和95%的醇濃度提取時,淫羊藿苷的重量較低;(3)70%乙醇濃度提取時,淫羊藿苷的總重量達到最高值。
4.3 從乙醇中提取淫羊藿總黃酮
圖3顯示:(1)50%乙醇提取時,取3倍量時淫羊藿總黃酮達到最高值;(2)70%乙醇提取時,隨著倍量的增加,總黃酮的重量呈線性速減;(3)20%和95%乙醇提取總黃酮時,總的重量較低;(4)用50%乙醇提取,總黃酮的總重量為最高。
4.4 水提物中提取總多糖
圖4顯示,70%濃度的乙醇提取時總多糖的損失較少,含量較高。95%乙醇提取后所得的水提物,黏性較大,得到總多糖的含量也較少。
[1]李時珍.本草綱目[M].第2冊.北京:人民衛(wèi)生出版社,1977.751.
[2]王昌林,李 昱,王粵新.淫羊藿及其有效成分的藥理研究概況[J].中醫(yī)中藥雜志,1988,23(3):183.
[3]雷載權,陳松育,高學敏.中藥學[M].上海:上海科學技術出版社,2001.288.
[4]閻愛榮,李愛軍.淫羊藿的現(xiàn)代藥理研究概述[J].傳統(tǒng)中藥研究,1999,2:62.
[5]蔡曼玲,季 暉,李 萍,等.5種淫羊藿黃酮類成分對體外成骨細胞的影響[J].中國天然藥物,2004,4:235.
[6]蔣麗君,夏新華.抗癡呆膠囊水提取工藝研究[J].湖南中醫(yī)學院學報,2000,20(2):26.
[7]國家藥典委員會.中華人民共和國藥典[S].2010年版一部.北京:中國醫(yī)藥科技出版社,307.
猜你喜歡
四川蠶業(yè)(2021年2期)2021-03-09 03:15:32
四川蠶業(yè)(2021年3期)2021-02-12 02:38:46
中成藥(2018年11期)2018-11-24 02:57:00
中成藥(2017年8期)2017-11-22 03:19:40
中成藥(2017年10期)2017-11-16 00:50:13
中成藥(2017年4期)2017-05-17 06:09:50
哈爾濱醫(yī)藥(2016年1期)2017-01-15 13:43:16
天然產(chǎn)物研究與開發(fā)(2016年11期)2016-06-15 20:29:17
湖南師范大學自然科學學報(2015年1期)2015-02-27 14:50:04
安徽醫(yī)藥(2014年12期)2014-03-20 13:15:15
