腦內補體系統與阿爾茨海默病①
2012-01-23 12:04:18李聞文中南大學湘雅醫學院檢驗系長沙410013
中國免疫學雜志 2012年12期
羅 浩 李聞文 周 軍 (中南大學湘雅醫學院檢驗系,長沙410013)
阿爾茨海默病(Alzheimer's Disease,AD)是臨床上最為常見的老年癡呆性疾病,臨床上表現為隱性起病、不可逆進行性發展的記憶減退,認知、語言功能障礙及人格的改變等,其病理特點為腦內Aβ聚集形成老年斑(Senile plaques,SP),神經元內tau蛋白異常改變形成神經纖維纏結(Neurofibrillary tangles,NFT),腦皮質及海馬膽堿能神經元及其突觸的大量丟失,皮質動脈出現血管淀粉樣變性[1]。目前,關于AD的病因及機制主要以遺傳因素、炎癥免疫學說、自由基學說為主,但這些學說解釋AD的發生發展有待于進一步完善,隨著研究的不斷深入,補體介導的免疫炎性學說越來越受到人們的重視[2]。
1 腦內補體系統
由于血腦屏障的存在,血細胞以及血漿蛋白等物質難以進入中樞神經系統。雖然血腦屏障并非能絕對阻止大分子物質進入腦內,但大多數補體蛋白不能完全透過血腦屏障,因此認為大腦是免疫豁免器官,其固有免疫反應受到極大限制[3]。近年研究發現,固有免疫的許多因子能在大腦中表達,腦內存在活躍的固有免疫反應,在維持膠質細胞內環境自穩,識別和清除侵襲性微生物、衰老細胞、過量神經遞質以及老化糖化蛋白等方面起到重要作用。如大腦受損后,所有補體成分都能在大腦內生成,伴隨疾病不同進展過程起著損傷或保護作用[4]。綜上所述,大腦細胞產生的補體蛋白可形成腦內局部固有防御體系,這些補體成分在大腦發育過程以及急慢性大腦損傷中均起到極為重要的調控作用。腦內細胞同時表達多種調控蛋白,如C1NH、H因子、備解素、MCP、P因子、DAF及其CD59等,這些蛋白為防止補體過度活化而導致自身組織損傷起到重要調控作用。由此可見,補體對中樞神經系統是防御,還是過度防御導致相關疾病,完全取決于補體蛋白在腦內的表達程度,一旦平衡失調,腦內結構及功能均出現紊亂。
目前認為,腦內補體一般分為三種來源:①膠質細胞見表1:包括星形膠質細胞、小膠質細胞、少突膠質細胞。Lévi-Strauss等[5]曾報道,小鼠星形膠質細胞能夠產生補體活化旁路途徑中的補體蛋白。近年研究表明,人類星形膠質細胞和小膠質細胞可產生經典途徑及其旁路途徑中的絕大多數的補體蛋白以及補體調節蛋白[6]。②神經元:腦組織原位雜交方法檢測證實 AD 腦組織神經元有 C1q、C2、C3、C4、C5、C6、C7、C8、C9mRNA 的表達[7]。③某些病理情況下[8],血腦屏障被破壞,從外周循環進入腦實質的中性粒細胞等能分泌補體,釋放炎癥因子,通過進一步的反饋作用促進腦內細胞補體自分泌。以上三條途徑均認為是參與腦內免疫炎性反應的結構基礎。
2 腦內補體激活途徑與調節作用
生理情況下,多數血清補體成分是以酶前體的形式存在。補體激活過程是一系列放大連鎖反應,包括三條途徑[9]:由抗原抗體復合物結合C1q啟動激活的經典途徑(Classical pathway,CP);由病原微生物等提供接觸表面,而從C3開始激活的旁路途徑(Alternative pathway,AP);由甘露聚糖結合凝集素(Mannose-binding lectin,MBL)結合細菌啟動的MBL途徑(MBL pathway,MBLP);最終通過C3的激活進入共同的終末途徑(Terminal pathway,TP)形成MAC[10]。近年有研究者用Aβ1-42處理大鼠海馬組織切片,發現C1q及其mRNA表達上調,表明大腦存在以C1q作為起始物的補體CP[11]。Strohmeyer等[12]在AD尸檢中發現,在大腦額葉皮質中補體B及激活產物Ba、Bb和FHL-1顯著高于對照組,而補體抑制物I因子和H因子與對照無顯著意義,證實腦內存在AP[12]。另外有學者認為MBLP也可能參與AD的病理過程。
炎癥反應導致大腦結構發生病理改變使其正常功能紊亂,而大腦具有自我調節機制可維持自穩。有研究報道,白細胞介素IL-1β、IL-6、干擾素INF-γ、腫瘤壞死因子TNF-α、H因子、ICAM、血清P蛋白、Aβ等多種細胞因子都可作為補體調節劑,其調節作用與上述因子產生的數量及其病理反應密切相關。C反應蛋白(CRP)是一種急性時相反應蛋白,它可作為一種補體系統激活劑,與相應配基結合,激活C1q從而激活經典的補體激活途徑[13]。在老年化相關疾病的研究中發現,AD是由于不斷積累的Aβ誘發補體成分活化,使膠質細胞上調,產生促炎癥因子、氧自由基等細胞毒性物質,最終大腦局部產生慢性炎癥反應。由于補體激活產物和補體調節蛋白失衡,如C1抑制物C1INH,C3轉化酶抑制物I因子、MCP、DAF等,該慢性炎癥反應將相應加重。活化的補體成分也能通過激活靜止的血管內皮細胞呈現多種生物學效應。
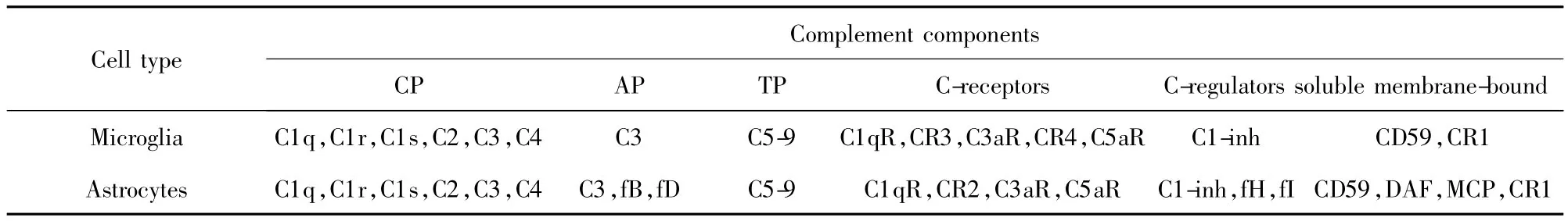
表1 人類小膠質細胞和星形膠質細胞所表達的補體蛋白及調節蛋白[8]Tab.1 Expression of C proteins and C-regulators by human astrocytes and microglia[8]
3 Aβ與補體在AD中的生物效應
使得AD致病的Aβ多為含有39~43個氨基酸殘基的多肽,分子量為4 kD。多肽構成β-片層結構,來源于淀粉樣前體蛋白(Amyloid precursor protein,APP)。APP基因在轉錄后由于不同的剪接產生10余種不同的mRNA和365~770個氨基酸殘基的蛋白質異構體,其中APP695、APP770是人腦內主要存在形式。Aβ是各種細胞APP正常代謝產物,中樞神經系統中神經元、星形膠質細胞、小膠質細胞、少突膠質細胞和內皮細胞均表達APP,APP在α-分泌酶、β-分泌酶和 γ-分泌酶的作用下裂解產生Aβ[14]。由于三種酶對Aβ的作用位點不一致,產生的Aβ對人體的生物學效應也不一致。研究證實,α-分泌酶裂解APP產生分泌性APP(APPsa),其可促進神經存活、抗神經興奮性氨基酸的毒副作用,在減少AD及神經退行性疾病中具有一定生理作用,即正常人大腦中存在Aβ,其可分泌至細胞外,具有可溶性,無積聚性及毒性作用。β-分泌酶主要裂解APP695中Met-595與 ASP-597之間膜外氨基端肽鍵,γ-分泌酶主要裂解Aβ39-43位跨膜羧基端任意肽鍵而產生分子大小不一的完整Aβ分子,其產生的Aβ具有很強的疏水性,因此認為β-分泌酶和γ-分泌酶的作用決定了Aβ的毒性作用[15]。綜上所述,正常情況下主要表現為α-分泌酶活性,而當大腦存在病理改變時β-分泌酶和γ-分泌酶含量將會升高或活性增強。研究表明,Aβ可引起膽堿能神經元損傷、神經細胞凋亡、氧應激和免疫炎癥反應,從而損傷神經細胞。但是可溶性的Aβ本身并無神經毒性,只有當β-折疊形成絲狀纖維聚集物變為不可溶性沉積物后則具有神經細胞毒性作用,研究發現,Aβ42、Aβ43毒性最強。亦有研究報道,可溶性、呈寡居狀態的Aβ寡聚體(Amyloid-beta oligomers,AβOs)也可能引起AD神經元功能失調,并可升高TNF-α、MCP-1、MIP-1α,促進 Aβ沉積,形成惡性循環的慢性免疫過程[16]。
研究表明,MAC有很明顯的神經毒性,其作用可溶解神經元[17]。凋亡的神經元周圍存在C1q和C5b-9等補體蛋白成分,提示AD患者神經元的凋亡很可能與經典補體途徑相關。據報道,C1q作為補體經典途徑活化的起始物,通過與Aβ結合,參與形成老年斑[18]。補體裂解產物 C3a、C4a、C5a 及其MAC具有過敏毒素作用,其中以C5a作用最強。AD在其不同的進展期,表現出不同的補體表達成分。在AD的早期,C1q、C4d和C3d的表達上調,而終末階段的C5b-9則缺失或不占主導作用;在疾病的發生發展過程中,NP及NFT周圍可觀察到較強的C1q、C4d、C3d以及部分C5b-9,提示補體經典途徑和C3在AD的主導作用[19]。對患有AD不同年齡段的尸檢研究也證實了這一主導作用。補體因子H近來倍受關注,其作為負性免疫炎性反應的調節因子,可通過抑制AP進而起到保護作用。有學者指出,聯合AD患者腦脊液中補體因子H和C3的檢測,可以作為評價 AD病理進程的指標[20]。Lukiw 等[21]研究發現,在 AD 中,miRNA-9、miRNA-125b、miRNA-146a和miRNA-155作為上調表達NF-κB的因素,可下調補體因子H的表達,即在AD進展中,補體因子H的表達受到抑制,從而加速了由AP引起的病理損害。
動物實驗發現,補體激活產物存在于大腦發育以及神經元形成過程中[22]。這些發現不但有助于了解大腦發育和神經元網絡形成的機理,同時也為補體系統在與CNS相關疾病中的神經退行性和神經保護性作用提供理論支撐。Webster等[23]通過離體實驗證實,C1q可增強小膠質細胞吞噬Aβ的作用,進而保護神經元免受損傷。也有研究表明,C1q能抑制Aβ和血清P蛋白所致的神經毒性。此外,C3也具有神經保護作用。Rogers等[24]發現C3b有助于C1R與Aβ結合,促進Aβ的清除,而另一補體激活物C3a可以誘導神經營養因子生成,防止門冬氨酸介導的神經毒性。進一步研究發現,C3a還具有抗炎作用。C5a作為過敏毒素最強的因子,仍有可能具有神經保護作用。體外誘導實驗發現,C5a能夠減少多種神經元的凋亡以及谷氨酸所誘發的神經毒性作用。C5a能夠促使小膠質細胞表達谷氨酸受體(GLT-1),進而促進了谷氨酸的吸收,通過阻斷谷氨酸的毒素作用保護神經系統[24]。Fonseca等[25]研究證實,C5a受體拮抗劑可抑制AP的激活,進而起到保護小鼠作用,同時也指出抑制補體活化能夠延緩AD小鼠病理進程。以上研究結果充分說明補體系統在腦內的復雜作用機制。
4 AD的免疫靶向治療和展望
補體受體拮抗劑和補體調節劑的使用,對于慢性神經退行性疾病以及急性神經系統疾病的治療具有不可低估的前景。研究顯示,AD的發病過程中,補體系統存在過度的活化。目前認為調節補體是最有希望干預AD發生發展的治療方法。藥物研究發現,痘病毒相關調節蛋白(VCP)、C5a受體拮抗劑PMX205、半枝蓮提取物B3-ps1等對AD有較好療效。VCP作為一種溫和的補體調節劑,可抑制補體系統的激活,減少Aβ的沉積改善大鼠的學習記憶能力。但目前為止,補體調節劑尚處于AD的動物研究階段[26]。此外,有研究報道,通過免疫方式減少腦內Aβ和過度磷酸化的Tau蛋白沉積,就能阻礙AD的發展[27,28]。目前進入臨床實驗的疫苗主要分為兩類:一類為通過主動免疫作用的疫苗,如CAD-106、ACC-001、ACI-24、UB-311、V-950 等;另一類為通過被動免疫作用的疫苗,如 Solanezumab(LY2062430)、Ponezumab(PF-04360365)等。
預計到2050年,全球將有1.06億AD患者,AD將成為全球致死性的神經退行性疾病。隨著人口老齡化日趨明顯,AD必將成為一個嚴重的社會問題。AD為一種不可逆進行性發展的腦損傷,除導致患者自身痛苦,也關系著家庭乃至整個社會的和諧,因此加大對AD的研究力度十分有必要。鑒于AD的藥物治療存在各種缺陷,就目前補體介導的炎性免疫反應機制提示,研制相應的補體激活劑、抑制劑,及其相關疫苗,在免疫炎性及補體效應的損傷階段進行干預,將有望能改善或治愈AD。
1 Querfurth H W,LaFerla F M.Alzheimer's disease[J].N Engl J Med,2010;362(4):329-344.
2 Broussard G J,Mytar J,Li R C et al.The role of inflammatory processes in Alzheimer's disease[J].Inflammopharmacology,2012;20(3):109-126.
3 Abbott N J,patabendige A A ,dolman D E et al.Structure and function of the blood-brain barrier[J].Neurobiol Dis,2010;37(1):13-25.
4 Veerhuis R,Nielsen H M,Tenner A J.Complement in the brain[J].Mol Immunol,2011;48(14):1592-1603.
5 Lévi-strauss M,Mallat M.Primary cultures of murine astrocytes produce C3 and factor B,two components of the alternative pathway of complement activation[J].J Immunol,1987;139(7):2361-2366.
6 Verkhratsky A,Parpura V.Recent advances in(patho)physiology of astroglia[J].Acta Pharmacol Sin,2010;31(9):1044-1054.
7 Shen Y,Li R ,McGeer E G et al.Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain[J].Brain Res,1997;769(2):391-395.
8 Gosselet F,Candela P,Cecchelli R et al.Role of the blood-brain barrier in Alzheimer's disease[J].Med Sci(Paris),2011;27(11):987-992.
9 Woodruff T M,Ager R R,Tenner A J et al.The role of the complement system and the activation fragment C5a in the central nervous system[J].Neuromolecular Med,2010;12(2):179-192.
10 Ricklin D ,Hajishengallis G,Yang K et al.Complement:a key system for immune surveillance and homeostasis[J].Nat Immunol,2010;11(9):785-797.
11 Fan R and Tenner A J.Complement C1q expression induced by Abeta in rat hippocampal organotypic slice cultures[J].Exp Neurol,2004;185(2):241-253.
12 Strohmeyer R,Shen Y and Rogers J.Detection of complement alternative pathway mRNA and proteins in the Alzheimer's disease brain[J].Brain Res Mol Brain Res,2000;81(1-2):7-18.
13 Bi B T,Lin H B,Cheng Y F et al.Promotion of β-amyloid production by C-reactive protein and its implications in the early pathogenesis of Alzheimer's disease[J].Neurochem Int,2011;60(3):257-266.
14 Haass C,Kaether C,Thinakaran G et al.Trafficking and proteolytic processing of APP[J].Cold Spring Harb Perspect Med,2012;2(5):a006270.
15 Petrlova J,Kálai T,Maezawa I et al.The influence of spin-labeled fluorene compounds on the assembly and toxicity of the aβ Peptide[J].PLoS One,2012;7(4):e35443.
16 Choi J G,Moon M,Kim H G.Gami-Chunghyuldan ameliorates memory impairment and neurodegeneration induced by intrahipp-ocampal Aβ 1-42 oligomer injection[J].Neurobiol Learn Mem,2011;96(2):306-314.
17 D'Andrea M R.Evidence that immunoglobulin-positive neurons in Alzheimer's disease are dying via the classical antibody-dependent complement pathway[J].Am J Alzheimer's Dis Other Demen,2005;20(3):144-150.
18 Benoit M E,Tenner A J.Complement protein C1q-mediated neuroprotection is correlated with regulation of neuronal gene and microRNA expression[J].J Neurosci,20112;31(9):3459-3469.
19 Zhou J,Fonseca M I,Pisalyaput K et al.Complement C3 and C4 expression in C1q sufficientand deficientmouse modelsof Alzheimer's disease[J].J Neurochem,2008;106(5):2080-2092.20 Wang Y,Hancock A M,Bradner J et al.Complement 3 and factor h in human cerebrospinal fluid in Parkinson's disease,Alzheimer's disease,and multiple-system atrophy[J].Am J Pathol,2011;178(4):1509-1516.
21 Lukiw W J,Surjyadipta B,Dua P et al.Common micro RNAs(miRNAs) target complement factor H(CFH) regulation in Alzheimer's disease(AD)and in age-related macular degeneration(AMD)[J].Int J Biochem Mol Biol,2012;3(1):105-116.
22 Fraser D A,Pisalyaput K and Tenner A J.C1q enhances microglial clearance of apoptotic neurons and neuronal blebs,and modulates subsequent inflammatory cytokine production[J].J Neurochem,2010;112(3):733-743.
23 Webster S D,Yang A J ,Margol L et al.Complement component C1q modulates the phagocytosis of Abeta by microglia[J].Exp Neurol,2000;161(1):127-138.
24 Rogers J,Li R,Mastroeni D et al.Peripheral clearance of amyloid beta peptide by complement C3-dependent adherence to erythrocytes[J].Neurobiol Aging,2006;27(12):1733-1739.
25 Fonseca M I,Chu S H,Berci A M et al.Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer's disease[J].J Neuroinflammation,2011;8(1):4-4.
26 Wu Y,Chen D F.Anti-complementary effect of polysaccharide B3-PS1 in Herba Scutellariae Barbatae(Scutellaria barbata) [J].Immunopharmacol Immunotoxicol,2009;31(4):696-701.
27 Wiessner C,Wiederhold K H,Tissot A C et al.The second-generation active Aβ immunotherapy CAD106 reduces amyloid accumulation in APP transgenic mice while minimizing potential side effects[J].J Neurosci,2011;31(25):9323-9331.
28 Siemers E R,Friedrich S,Dean R A et al.Safety and changes in plasma and cerebrospinal fluid amyloid beta after a single administration of an amyloid beta monoclonal antibody in subjects with Alzheimer disease[J].Clin Neuropharmacol,2010;33(2):67-73.
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
中學生數理化·七年級數學人教版(2019年10期)2019-11-25 07:33:58
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
中學生數理化·高一版(2018年9期)2018-10-09 06:46:50
湖南教育·C版(2018年3期)2018-06-05 16:54:36
汽車工程學報(2017年2期)2017-07-05 08:13:02
中國衛生(2016年3期)2016-11-12 13:23:26
