高效液相色譜法檢測多種動物組織中氟喹諾酮類藥物殘留量的研究
2012-10-09 06:11:18張玉潔王鶴佳
中國獸藥雜志 2012年7期
關(guān)鍵詞:檢測
張玉潔,李 倩,汪 霞,王鶴佳,薛 毅,仲 鋒
(1.中國獸醫(yī)藥品監(jiān)察所,北京 100081;2.中國動物疫病預(yù)防控制中心,北京 100026)
氟喹諾酮類藥物(FQs)自20世紀(jì)80年代初問世以來,相繼被合成了許多廣譜、高效、低毒的廣譜殺菌性抗菌藥,主要的藥物有諾氟沙星、二氟沙星、恩諾沙星、環(huán)丙沙星、沙拉沙星、達氟沙星等。它們的基本作用相似,對革蘭氏陽性菌、陰性菌、支原體、某些厭氧菌均有效。本類藥物的毒副作用小,安全范圍較大。但本類藥物對動物骨骼生長發(fā)育有不良影響,禁用于幼齡動物和孕畜,同時對胃腸道、中樞神經(jīng)、肝細胞均有一定的危害[1]。隨著氟喹諾酮類藥物在獸醫(yī)臨床上的廣泛應(yīng)用以及耐藥性的產(chǎn)生,F(xiàn)Qs在可食性動物組織中的殘留問題已引起人們的高度關(guān)注。
農(nóng)業(yè)部第235號公告[2]對恩諾沙星、環(huán)丙沙星、沙拉沙星、達氟沙星及二氟沙星在多種動物組織中的最高殘留限量(MRL)進行了規(guī)定。而現(xiàn)在所用標(biāo)準(zhǔn)農(nóng)業(yè)部第236號公告《動物性食品中恩諾沙星和環(huán)丙沙星殘留檢測方法—高效液相色譜法》[3]和農(nóng)業(yè)部第1025號公告—14—2008《動物性食品中氟喹諾酮類藥物殘留檢測高效液相色譜法》[4]在檢測藥物數(shù)量、動物品種及動物組織覆蓋面方面均滿足不了實際檢測的要求。同時,沙拉沙星在雞的肌肉組織中的最高殘留限量為10μg/kg,上述兩個標(biāo)準(zhǔn)的定量限也不能滿足限量要求。因此,本研究在以上兩個檢測方法的基礎(chǔ)上做進一步改進,增加了檢測藥物、動物品種及動物組織,降低了沙拉沙星定量限,以便更好地滿足實際檢測工作的需求。
1 材料與方法
1.1 儀器設(shè)備、試劑及對照品 Agilent 1100高效液相色譜儀(配熒光檢測器);SPE柱:Bond Elut C18固相萃取柱(100 mg/1mL);固相萃取裝置;pH計;高速冷凍離心機;Milli-Q超純水儀。
環(huán)丙沙星、恩諾沙星、二氟沙星對照品,純度≥99.0%,由中國獸醫(yī)藥品監(jiān)察所提供;達氟沙星對照品,純度99.8%,由Sigma-Aldrich公司提供;沙拉沙星對照品,純度 95.0%,由Dr.Ehrenstorfer公司提供。甲醇、乙腈為色譜純,磷酸、氫氧化鈉、三乙胺、磷酸二氫鉀、檸檬酸、乙酸銨均為分析純。試驗用水為符合國家標(biāo)準(zhǔn)的一級水。
1.2 溶液配制 5.0 mol/L氫氧化鈉溶液:取氫氧化鈉飽和液28 mL,加水稀釋至100 mL,混勻。0.03 mol/L氫氧化鈉溶液:取5.0 mol/L氫氧化鈉液0.6 mL,加水稀釋至 100 mL,混勻。0.05 mol/L磷酸/三乙胺溶液:取濃磷酸3.4 mL,用水稀釋至1000 mL,用三乙胺調(diào)pH值至2.4。磷酸鹽緩沖溶液(用于肌肉、脂肪組織):取磷酸二氫鉀6.8 g,加水溶解并稀釋至500mL,用5.0mol/L氫氧化鈉溶液調(diào)節(jié)pH值至7.0。磷酸鹽溶液(用于肝臟、腎臟組織):取磷酸二氫鉀6.8 g,加水溶解并稀釋至500mL,混勻。洗脫液:取乙腈20 mL,用0.05mol/L磷酸/三乙胺溶液稀釋至100 mL,混勻。
5種FQs標(biāo)準(zhǔn)儲備液:分別取達氟沙星對照品約10 mg,環(huán)丙沙星、恩諾沙星、沙拉沙星和二氟沙星對照品各約50 mg,精密稱定于50 mL量瓶中,用0.03 mol/L氫氧化鈉溶液溶解并稀釋成濃度為0.2 mg/mL(達氟沙星)和1 mg/mL(環(huán)丙沙星、恩諾沙星、沙拉沙星、二氟沙星)的標(biāo)準(zhǔn)儲備液。置2~8℃冰箱中保存,有效期為3個月。5種FQs標(biāo)準(zhǔn)工作液:準(zhǔn)確量取適量標(biāo)準(zhǔn)儲備液,用乙腈稀釋成達氟沙星(2μg/mL和0.2μg/mL)、環(huán)丙沙星、恩諾沙星、沙拉沙星和二氟沙星(10μg/mL和1μg/mL)的標(biāo)準(zhǔn)工作液。置2~8℃冰箱中保存,有效期為1個月。
1.3 試驗方法
1.3.1 液相色譜條件 色譜柱:C18250 mm×4.6 mm(i.d),粒徑 5 μm;流動相:0.05 mol/L磷酸/三乙胺溶液+乙腈(85+15,V/V),使用前經(jīng)微孔濾膜過濾(測肌肉、脂肪、肝臟組織用);檸檬酸/乙酸銨溶液+乙腈(85+15,V/V),使用前經(jīng)微孔濾膜過濾(測腎臟組織時用);流速:0.8 mL/min;檢測波長:激發(fā)波長280 nm;發(fā)射波長450 nm;柱溫30℃;進樣量40μL。
1.3.2 標(biāo)準(zhǔn)曲線及線性范圍的測定 準(zhǔn)確量取適量FQs標(biāo)準(zhǔn)工作液,用流動相稀釋成濃度分別為0.5、1、5、10、50、100 μg/L(以恩諾沙星為例)的對照溶液,供高效液相色譜分析。以各色譜峰峰面積為縱坐標(biāo),對照溶液濃度為橫坐標(biāo),繪制標(biāo)準(zhǔn)曲線。1.3.3 樣品前處理 取(2 ± 0.05)g試料,置50 mL離心管中,肌肉、脂肪試樣加磷酸鹽緩沖溶液10.0 mL,肝臟、腎臟試樣加磷酸鹽溶液 20.0 mL,渦旋混勻,中速振蕩 5 min。10000 r/min離心10 min,取上清液于另一離心管中。肌肉、脂肪試樣殘渣中再加磷酸鹽緩沖溶液10.0 mL;肝臟、腎臟試樣殘渣中再加磷酸鹽溶液20.0 mL,重復(fù)提取一遍。合并兩次上清液,充分混勻,備用。
取腎臟試樣上清液10 mL于另一50 mL離心管中,加5 mL正己烷,中速振蕩5 min。5000 r/min離心5 min,取下層清液,混勻,備用。
固相萃取柱依次用甲醇、水各2 mL預(yù)洗。取備用液3.0 mL過柱,超純水1 mL淋洗,擠干。用洗脫液1.0 mL洗脫,擠干,收集洗脫液。經(jīng)濾膜過濾后作為試樣溶液,供高效液相色譜法測定。
1.3.4 檢測限及定量限 分別取五種動物各自肌肉、脂肪、肝臟、腎臟4種組織的空白樣品進行添加回收實驗,按照上述樣品前處理方法處理,進HPLC分析。以S/N≥10,且在該添加水平的回收率和變異系數(shù)均滿足殘留分析要求的最小濃度作為定量限(LOQ),以S/N=3作為藥物的檢測限(LOD)。
1.3.5 準(zhǔn)確度和精密度 采用標(biāo)準(zhǔn)添加法,在5種動物的肌肉、脂肪空白組織中添加3個不同濃度(5μg/kg、100μg/kg和200μg/kg)(以恩諾沙星為例)的FQs標(biāo)準(zhǔn)溶液進行回收率測定,每個濃度設(shè)定5個平行樣品,同時進行空白試驗,重復(fù)3次,求每個樣品的回收率和批內(nèi)、批間相對標(biāo)準(zhǔn)偏差(RSD)。按照相同方法,在5種動物的肝臟、腎臟空白組織中添加3個不同濃度(10μg/kg、100 μg/kg和200μg/kg)(以恩諾沙星為例)的FQs標(biāo)準(zhǔn)溶液,求每個樣品的回收率和批內(nèi)、批間RSD。
2 試驗結(jié)果
2.1 標(biāo)準(zhǔn)曲線及線性范圍 達氟沙星在0.1~20 μg/L濃度范圍內(nèi),其他4種FQs藥物在0.5~100 μg/L濃度范圍內(nèi)呈現(xiàn)良好的線性關(guān)系,其曲線方程和相關(guān)系數(shù)(R2)見表1所示。

表1 5種FQs藥物的回歸方程及相關(guān)系數(shù)
2.2 檢測限及定量限 空白試料按1.3.3步驟處理后,測定結(jié)果表明:在相應(yīng)的保留時間處,大部分空白試料對所測分析物無干擾;少部分空白組織在藥物色譜峰保留時間處有較小干擾,可調(diào)整流動相比例避開干擾物質(zhì)或者在回收率計算中減去空白。圖1~圖3給出了標(biāo)準(zhǔn)對照液、牛腎臟空白樣品及組織添加樣品的色譜圖。
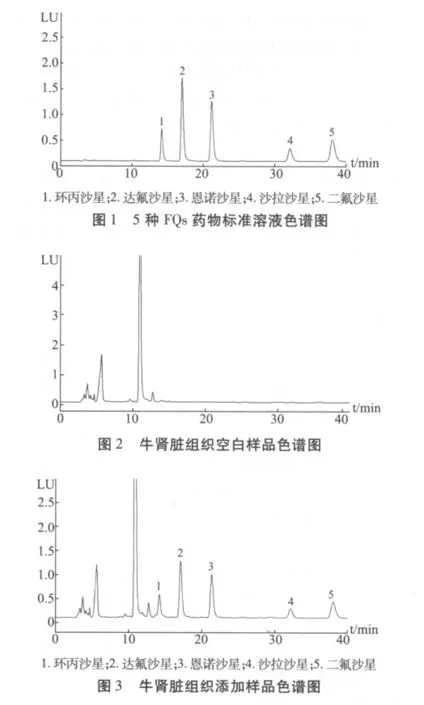
在豬、雞、牛、羊、兔的肌肉、脂肪組織中添加5種FQs藥物的標(biāo)準(zhǔn)溶液,使其在組織中的添加濃度分別為1、2.5 和 5 μg/kg(達氟沙星為 0.2、0.5和1μg/kg)。按照1.3.3提取凈化方法處理后,經(jīng)HPLC檢測。測定結(jié)果顯示,添加濃度為2.5μg/kg時,藥物的信噪比(S/N)約等于3,添加濃度為5μg/kg時,信噪比均大于10,為此,確定環(huán)丙沙星、恩諾沙星、沙拉沙星和二氟沙星在豬、雞、牛、羊、兔的肌肉、脂肪組織中的檢測限為2.5μg/kg,定量限為5μg/kg;達氟沙星的檢測限為0.5μg/kg,定量限為1 μg/kg。
按與上述肌肉、脂肪組織相同的處理和分析,確定環(huán)丙沙星、恩諾沙星、沙拉沙星和二氟沙星在豬、雞、牛、羊、兔的肝臟、腎臟組織中的檢測限為5μg/kg,定量限為10μg/kg;達氟沙星的檢測限為1 μg/kg,定量限為2 μg/kg。
2.3 準(zhǔn)確度和精密度 豬、雞、牛、羊、兔各四種組織中三個濃度FQs藥物添加回收試驗的回收率、批內(nèi)RSD和批間RSD結(jié)果見表2。
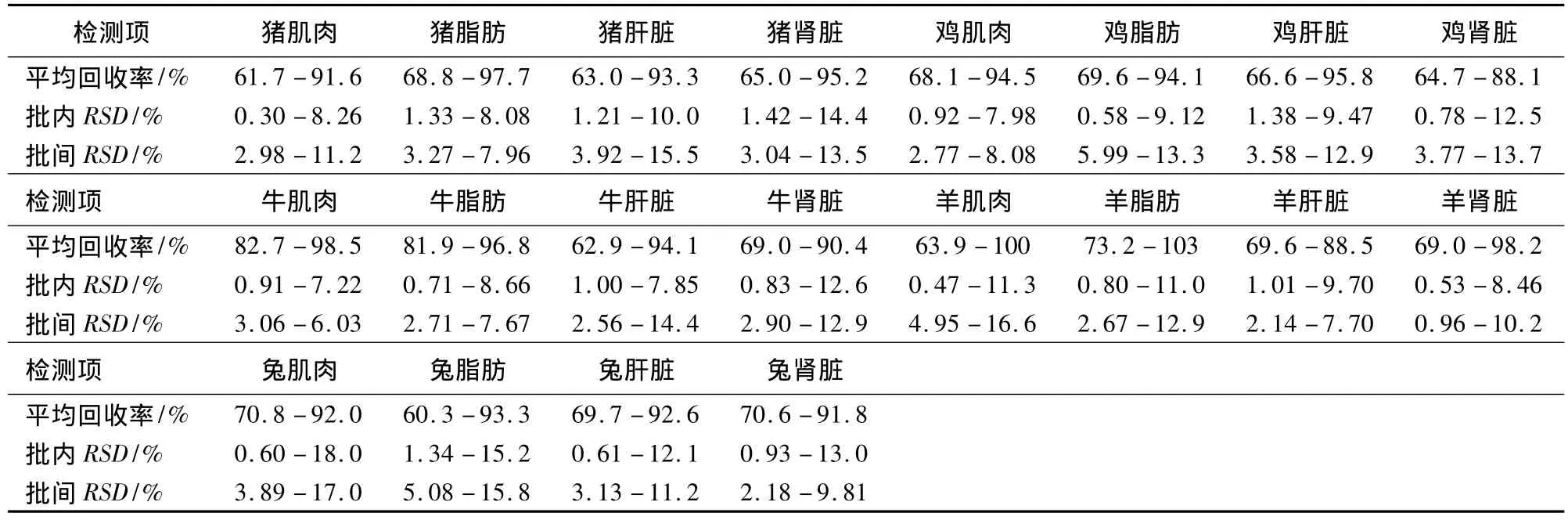
表2 5種FQs藥物準(zhǔn)確度及精密度試驗數(shù)據(jù)(n=5)
由表2數(shù)據(jù)可知,該方法在五種動物共20種組織中的平均回收率為62% ~103%;批內(nèi)變異系數(shù)為0.30% ~18%,批間變異系數(shù)為1.0% ~17%。方法準(zhǔn)確度、精密度良好。
3 討論
3.1 色譜條件的優(yōu)化 由于在測定腎臟組織時雜質(zhì)干擾較多,尤其是在牛的腎臟組織測定時,用原流動相 0.05 mol/L磷酸溶液/三乙胺 -乙腈(85∶15,V/V),在被測物出峰時有較強的干擾,改變原流動相的比例也無法使其分離。因此在腎臟組織的提取過程中,增加了正己烷除脂的步驟,并用檸檬酸/乙酸銨溶液-乙腈(85∶15,V/V)作為流動相,試驗結(jié)果顯示(見圖1-3),經(jīng)除脂和更換流動相后,在藥物色譜峰保留時間處基本不再有較強的雜質(zhì)峰干擾。最終確定在測定腎臟組織時,用檸檬酸/乙酸銨溶液 -乙腈(85∶15,V/V)作為流動相;在測定其它組織時,仍使用原流動相0.05mol/L磷酸溶液/三乙胺-乙腈(85∶15,V/V)。
3.2 提取條件的優(yōu)化 農(nóng)業(yè)部第236號公告《動物性食品中恩諾沙星和環(huán)丙沙星殘留檢測方法—高效液相色譜法》[3]和農(nóng)業(yè)部第1025號公告—14—2008《動物性食品中氟喹諾酮類藥物殘留檢測高效液相色譜法》[4]中對雞肌肉、脂肪、肝臟、腎臟四種組織均用10 mL提取液提取兩次,經(jīng)對更多的動物組織的試驗摸索,新方法對肝臟、腎臟組織采用了20 mL提取液提取兩次,不僅對殘留的藥物提取得更充分,同時降低了單位體積提取液中的雜質(zhì)量。
3.3 凈化條件的優(yōu)化 通過對200 mg/3 mL、500 mg/6 mL以及100 mg/1 mL等多種不同規(guī)格C18固相萃取柱的提取、凈化實驗,結(jié)果表明:規(guī)格大的C18固相萃取柱,雖然可過更多體積的提取液,進一步提高方法的靈敏度,但需要增加更多的有機溶劑和提取步驟,如氮吹或旋轉(zhuǎn)蒸發(fā)等。而用原標(biāo)準(zhǔn)中的C18 100 mg/1 mL固相萃取柱,雖然方法靈敏度沒有用大規(guī)格C18固相萃取柱高,但也足以滿足235號公告中的限量要求。為此,該方法仍沿用吸附能力既能滿足限量檢測要求,同時又能充分洗脫的100 mg/1 mL C18固相萃取柱。
為了在滿足方法定量限要求的前提下進一步簡化操作步驟、減少雜質(zhì)干擾,該試驗取3.0 mL提取液過固相萃取柱。對比農(nóng)業(yè)部公告第236號《動物性食品中恩諾沙星和環(huán)丙沙星殘留檢測方法—高效液相色譜法》中的“取適量備用液過柱”和農(nóng)業(yè)部1025號公告—14—2008《動物性食品中氟喹諾酮類藥物殘留檢測高效液相色譜法》中“取5.0 mL備用液過柱”,取3.0 mL提取液過柱不僅滿足了定量限的要求,而且簡化了操作步驟,更重要的是減少了組織中的雜質(zhì)干擾,避免了固相萃取柱的堵塞,加快了凈化步驟,也縮短了整個樣品的前處理時間。
相對于現(xiàn)有標(biāo)準(zhǔn)方法,本方法提高了樣品提取時的離心速度,其目的是盡量減少懸浮于提取液中的組織雜質(zhì),避免過固相萃取柱時造成柱子堵塞。這一改變在處理肝臟、腎臟時效果更加明顯。
相對于其他三種組織,經(jīng)磷酸鹽溶液提取并將兩次提取液合并后,腎臟組織的雜質(zhì)干擾更多,不僅過SPE柱的過程更為困難,并且在環(huán)丙沙星出峰的保留時間處有明顯干擾。因此,在20mL提取液合并后,取出10mL加入5mL正己烷除脂,振蕩、離心后取下層清液過SPE柱。經(jīng)此改進后,腎臟組織的前處理過程更為順利,HPLC分析結(jié)果也顯示雜質(zhì)干擾大大減少。
綜上所述,該方法操作簡單,靈敏度高,準(zhǔn)確度和精密度好,藥物和動物組織覆蓋范圍廣,可以很好地滿足實際檢測工作的需要。
[1] 李俊鎖,邱月明,王 超.獸藥殘留分析[M].上海:上海科學(xué)技術(shù)出版社,2O02:257-262.
[2] 中華人民共和國農(nóng)業(yè)部公告第235號《動物性食品中獸藥最高殘留限量》[Z].2002.
[3] 中華人民共和國農(nóng)業(yè)部公告第236號《動物性食品中恩諾沙星和環(huán)丙沙星殘留檢測方法—高效液相色譜法》[Z].2003.
[4] 中華人民共和國農(nóng)業(yè)部公告1025號—14—2008《動物性食品中氟喹諾酮類藥物殘留檢測高效液相色譜法》[Z].2008.
猜你喜歡
中國設(shè)備工程(2022年12期)2022-07-11 04:33:00
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:36
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:34
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:50
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:48
