鋁鎵咔咯配合物的發(fā)光性質、堆積效應與軸向配位作用
2013-09-15 01:41:10霜陳張松金李偉英邵文莉
無機化學學報 2013年1期
何 霜陳 歡 張松金 李偉英 邵文莉
章 浩1 王 惠2 計亮年2,3 劉海洋*,1
(1華南理工大學化學與化工學院,廣州 510640)
(2中山大學光電材料與技術國家重點實驗室,廣州 510275)
(3中山大學生物無機與合成化學教育部重點實驗室,廣州 510275)
咔咯大環(huán)金屬配合物是當今卟啉化學的重要研究課題[1-5]。第三主族元素的咔咯配合物有獨特的結構與光物理特性[6],其研究近幾年受到人們的關注。Ghosh和Brothers采用密度泛函理論研究硼咔咯和銦咔咯配合物[7]。發(fā)現(xiàn)硼咔咯配合物中的B原子可以單體或雙體形式鍵入咔咯中心,并存在順式和反式兩種構型;銦咔咯則存在核心擴展效應(coreexpansion effects),與鎵咔咯相比,相鄰2個N原子之間的距離明顯變大。鋁和鎵咔咯具有特殊的光物理性能,在分子探針、腫瘤診斷與治療方面顯示出良好的應用前景。如鎵咔咯具有很好的靶向性,與特定蛋白結合后能夠殺死癌細胞而不損傷正常組織[8-10]。水溶性的鋁和鎵咔咯[11]能夠增強魯米諾(Luminol)體系的化學發(fā)光,為新型生物探針的研究提供了新的途徑。碘化后的鋁咔咯有著比其他的鋁咔咯更長的三重態(tài)壽命,可應用于光動力治療領域[12]。
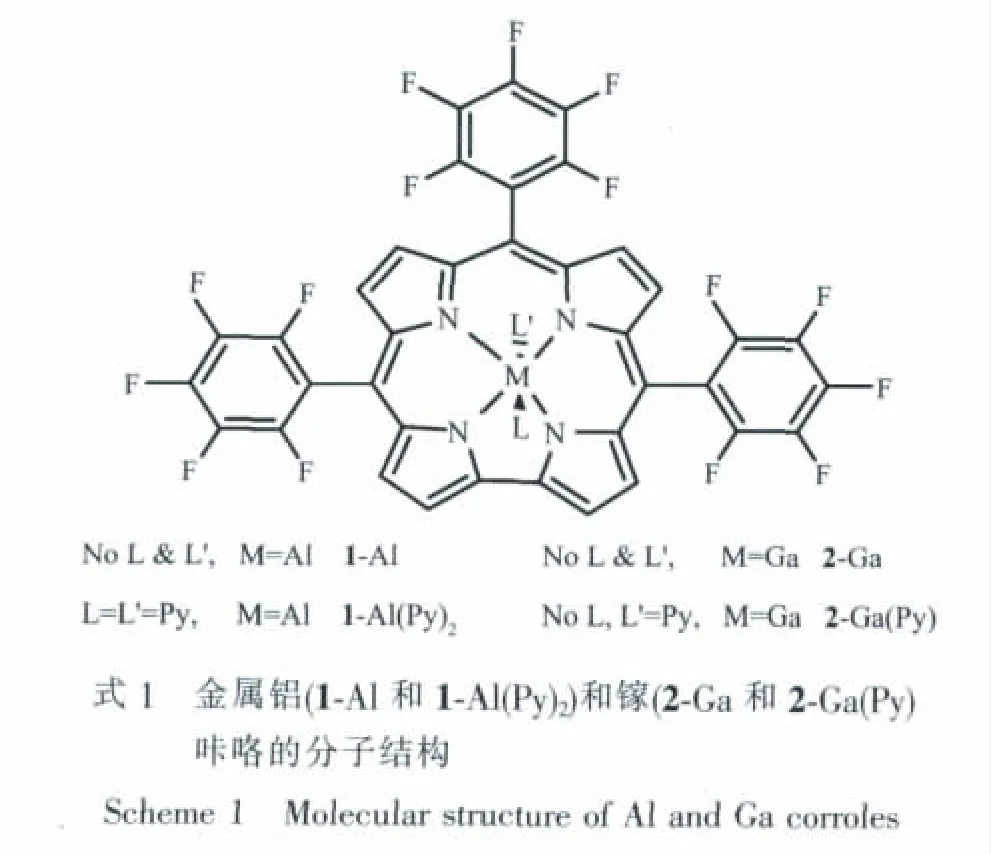
盡管第三主族元素的咔咯配合物研究已取得了相當的進展,但配合物在溶液中的基礎性質仍然有待深入。 本文合成了 5,10,15-三(五氟苯基)咔咯的2種第三主族金屬鋁、鎵配合物(見式1),測定了甲苯溶液中4種配合物的電子光譜、熒光光譜、熒光壽命以及熒光量子產率等發(fā)光性質;探討了配合物在二氯甲烷溶液中的聚集行為及軸向配位作用。
1 實驗部分
1.1 試劑和儀器
2 mol·L-1的三甲基鋁的甲苯溶液 (西亞試劑);氯化鎵(>99%,Sigma);咪唑(>99%,Sigma 公司);4-甲基咪唑(>99%,西亞試劑)。H3(tpfc)的合成參照Gross組的參考文獻[13];甲苯、吡啶、四氫呋喃、乙腈、丙酮、乙醚、N,N-二甲基甲酰胺(分析純,廣州試劑廠);二氯甲烷、正己烷、甲醇、乙酸乙酯、乙醇(分析純,國藥集團化學試劑有限公司);以上試劑除甲苯作為反應試劑進行純化干燥處理外,其余試劑均直接使用。
電子吸收光譜和光度值用U-3900H紫外可見分光光度計(日本-Hitachi)測定,熒光發(fā)射光譜采用F-4500熒光分光光度計(日本-Hitachi),質譜采用德國布魯克Esquire HCT PLUS型液相色譜-質譜聯(lián)用儀,核磁NMR采用德國布魯克AVANCE Digital 400 MHz超導核磁共振譜儀,熒光壽命測定采用英國EDINBURGH INSTRUMENTS LTD公司的FLSP920組合式熒光壽命和穩(wěn)態(tài)光譜儀,PG401SH/DFG2-10 ps激光器。
H3(tpfc)的合成參照Gross組的文獻[13],金屬鋁和鎵金屬配合物合成分別參照Mahammed和Gross[14]和Bendix等[15]文獻,經過改良合成步驟,適當處理。2-Ga的合成:稱取0.02 g的帶軸向配體的咔咯鎵配合物(2-Ga(Py)),溶解于20 mL二氯甲烷中;然后把2-Ga(Py)二氯甲烷溶液轉入分液漏斗中,并向其中加入質量分數為1%的鹽酸溶液40 mL;振蕩、搖勻分液漏斗中的溶液,靜置、分層,分出有機相;繼續(xù)以上方法把2-Ga(Py)二氯甲烷溶液酸洗2次,把得到的有機相再轉入分液漏斗中,并加入40 mL NaHCO3溶液(5%),振蕩、靜置、分液,得到的有機相用無水碳酸鈉干燥2 h,過濾、旋干溶劑,得到無軸向配體的鎵咔咯(2-Ga)的粗產品。把上述無軸向配體的鎵咔咯(2-Ga)的粗產品用柱層析分離(VCH2Cl2∶VCH3OH=100∶1),收集暗紅色一段主要產物,蒸去溶劑,真空干燥后得純凈的2-Ga,產率為90%。
化合物表征 1-Al:1H NMR(CDCl3,400 MHz),δ:9.27(d,2H,β-H),8.88(d,2H,β-H),8.80(d,2H,β-H),8.68(d,2H,β-H)。19F NMR(CDCl3,400 MHz),δ:-137.75(s,6F,ortho-F),-153.90(d,3F,para-F),-162.51(d,6F,meta-F)。APCI-MS:m/z 820.06(M+,100%)。 UV-Vis(toluene),λmax(ε×10-4):401(4.18),423(21.57),575(1.70),594(2.02)。 1-Al(Py)2:1H NMR(CDCl3,400 MHz),δ:9.26(d,2H,β-H),8.87(d,2H,β-H),8.82(d,2H,β-H),8.67(d,2H,β-H),6.70(t,2H,para-H of pyridine),5.92(unresolved t,4H,meta-H of pyridine),5.29(unresolved d,4H,ortho-Hof pyridine)。19FNMR(CDCl3,400MHz),δ: -138.04 (m,6F,ortho-F), -154.0(m,3F,para-F),-162.56(m,6F,meta-F)。 APCI-MS:m/z 820.09(M+-2pyridine,100%)。 UV-Vis(toluene),λmax(ε×10-4):400(5.12),423(27.96),575(2.14),594(2.48)。 2-Ga 表征:1H NMR(CDCl3,400 MHz),δ:9.14(d,2H,β-H),8.82(d,2H,β-H),8.76(d,2H,β-H),8.61(d,2H,β-H)。19F NMR(CDCl3,400 MHz),δ:-137.73(s,6F,ortho-F),-153.86(dt,3F,para-F),-162.43(s,6F,meta-F)。APCI-MS:m/z 863.1(M+,100%)。 UV-Vis(toluene),λmax(ε×10-4):402(5.32),424(25.13),571(1.82),596(2.34)。2-Ga(Py)表征 :1H NMR(CDCl3,400 MHz),δ:9.13(d,2H,β-H),8.76(d,2H,β-H),8.67(d,2H,β-H),8.63(d,2H,β-H),6.26(t,1H,para-H of pyridine),5.58(unresolved t,2H,meta-H of pyridine),2.81(unresolved d,2H,ortho-H of pyridine)。19F NMR(CDCl3,400 MHz),δ:-137.82(d,6F,ortho-F),-153.85(m,3F,para-F),-162.40(s,6F,meta-F)。 APCI-MS:m/z 863.1(M+-pyridine,100%)。UV-Vis(toluene),λmax(ε*10-4):401(5.16),424(28.85),571(1.99),596(2.55)。
1.2 實驗過程
電子吸收光譜測定在室溫下,不同種溶劑,在UV-Vis光譜儀上分別測定各溶液的紫外可見吸收光譜,濃度為5μmol·L-1。在穩(wěn)態(tài)熒光光譜儀上,分別測定各溶液的穩(wěn)態(tài)熒光光譜,濃度為5μmol·L-1。測試條件:室溫,樣品池為1 cm×1 cm×4 cm可密封石英皿,激發(fā)波長的狹縫寬度為5 nm,記錄波長范圍為 560~750 nm。
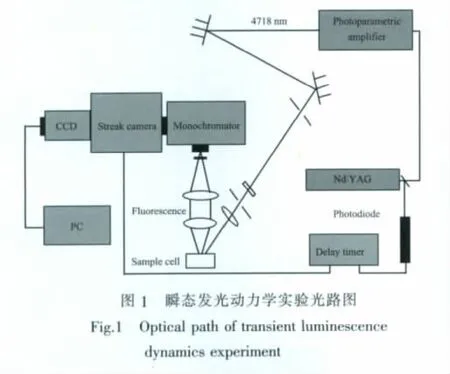
瞬態(tài)發(fā)光動力學過程使用圖1所示的光路進行測量,光源是一臺由立陶宛EKSPLA公司制造的PG401SH/DFG2-10 ps激光器,具體參數為:波長調諧范圍0.2~10μm,脈沖寬度25 ps,重復頻率10 Hz,單脈沖能量200μJ。實驗時用560 nm波長的光作為激發(fā)光,通過準直聚焦到樣品。樣品所發(fā)熒光由一組大口徑透鏡組合收集起來,光通過一臺反射式單色儀后由條紋相機 (HammatsuC1587)和CCD(C4742-95)進行記錄。將實驗所測數據Fm(t)和儀器響應函數E(t)進行解卷積得到樣品的瞬態(tài)發(fā)光衰減曲線和樣品發(fā)光壽命。所有實驗數據均在室溫(25±1)℃下測得。利用UV-Vis研究在溶液體系1-Al、1-Al(Py)2、2-Ga、2-Ga(Py)在 DCM 溶液中的聚集行為,以及1-Al、2-Ga與配體咪唑、4-甲基咪唑、吡啶的軸向配位作用。
2 結果與討論
2.1 電子光譜
金屬鋁和鎵的電子吸收光譜都表現(xiàn)出特征的Soret帶(420 nm 左右)和 Q 帶(550~650 nm 之間)的吸收峰。從圖2中可以看出,1-Al、2-Ga與1-Al(Py)2、2-Ga(Py)的電子吸收光譜類似,不帶配體吡啶的1-Al和2-Ga的Soret帶吸收峰較后兩者發(fā)生小幅度的藍移(2~4 nm),配合物的吸收強度則存在明顯差異,摩爾消光系數也不同,在軸向配體吡啶的作用下,1-Al(Py)2279 600,ε/(L·mol-1·cm-1)和 2-Ga(Py)(288 500)的消光系數比不帶軸向配體的1-Al(215 700)和 2-Ga(251 300)略大。
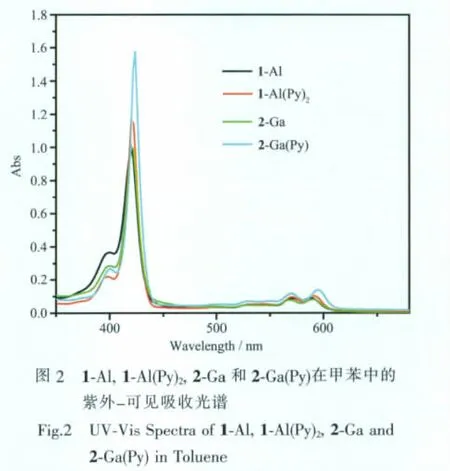
在甲苯、二氯甲烷、甲醇、乙醇、四氫呋喃、正己烷等幾種溶劑的作用下 (表1),由于帶軸向配體的1-Al(Py)2和2-Ga(Py)在溶劑中的解離作用,1-Al和1-Al(Py)2在同一種溶劑中的吸收峰偏移不大。在不同溶劑作用下,1-Al(Py)2在最大吸收峰位置偏移達8 nm(乙醇中為424 nm,丙酮為416 nm)。其他的配合物也類似。1-Al(Py)2、2-Ga(Py),相比不帶軸向配體吡啶的1-Al和2-Ga的Soret帶和Q帶的最大吸收峰發(fā)生略微藍移(2~4 nm)。金屬咔咯脫去軸向配體供電子基團吡啶后,1-Al和2-Ga中的電子分布和平面性發(fā)生改變,其電子吸收光譜發(fā)生一定程度的藍移,而軸向配體的缺失對金屬咔咯的共軛結構影響不大,因而吸收峰位置略微偏移。在不同的介電常數、不同極性的溶劑作用下,金屬咔咯的電子吸收光譜吸收峰吸收峰位置發(fā)生偏移,吸收強度也存在差異。
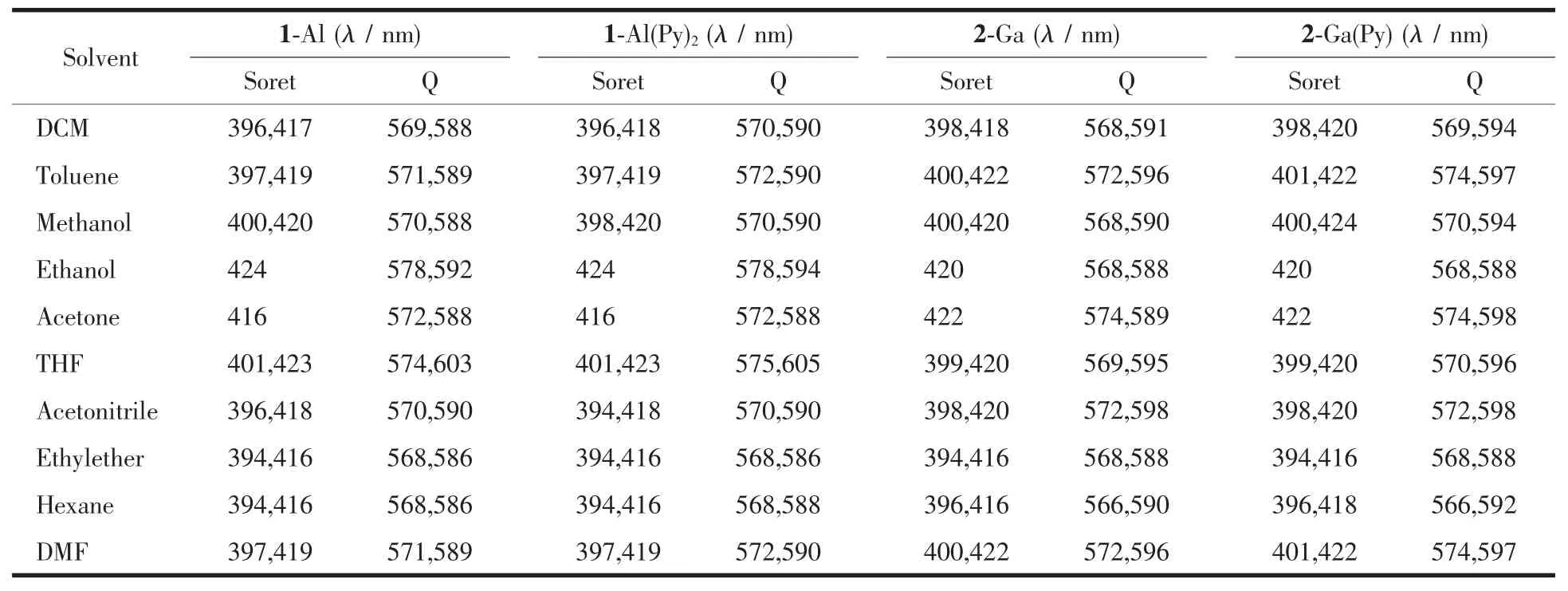
表1 鋁和鎵咔咯配合物在不同溶劑中的紫外-可見光譜數據Table 1 UV-Vis spectra data of aluminum and gallium corroles in different solvents
2.2 發(fā)光性質
鋁和鎵咔咯具有典型的雙熒光性質[6b],從第二單重激發(fā)態(tài)到基態(tài)(S2-S0)和第一單重激發(fā)態(tài)到基態(tài)(S1-S0)的熒光發(fā)射,通常需要采用熒光上轉化技術才能觀測到S2-S0發(fā)射峰。圖3是在甲苯溶劑中,1-Al、2-Ga、1-Al(Py)2、2-Ga(Py)的穩(wěn)態(tài)熒光發(fā)射光譜。在相同濃度下,激發(fā)波長為420 nm時,最大發(fā)射峰位置存在一定的偏移,發(fā)射光譜的強度明顯大于550 nm處發(fā)射光譜。
在不同溶劑中,1-Al(Py)2熒光發(fā)射光譜(圖4)的最大的發(fā)射峰位置與熒光強度存在明顯差異。激發(fā)波長為420 nm時,同一種溶劑對應的熒光發(fā)光強度明顯比在550 nm激發(fā)時大,同時,不同溶劑對熒光發(fā)光強度和發(fā)射峰的位置產生影響,弱極性的溶劑產生的熒光強度大于極性較大的溶劑,同時,部分溶劑使得熒光發(fā)射峰發(fā)生略微紅移。結合Franck-Condon原理及溶劑弛豫得到溶劑對分子熒光影響,溶劑取向作用影響非輻射弛豫的能量損失,而這與溶劑的介電常數ε和折射率n有關[16]。結合溶劑的極性、折光率等參數,溶劑對熒光光譜的影響,有待深入的探討。
表 2列出了不同溶劑中 1-Al、1-Al(Py)2、2-Ga和2-Ga(Py)在420 nm和550 nm激發(fā)下發(fā)射光譜的最大發(fā)射峰和熒光強度。1-Al較1-Al(Py)2,2-Ga較2-Ga(Py)發(fā)射峰在軸向配體吡啶的作用下發(fā)生一定程度的偏移。在同一種溶劑的作用下,激發(fā)波長為420 nm和550 nm時,Soret帶激發(fā)的發(fā)射峰強度明顯大于Q帶。
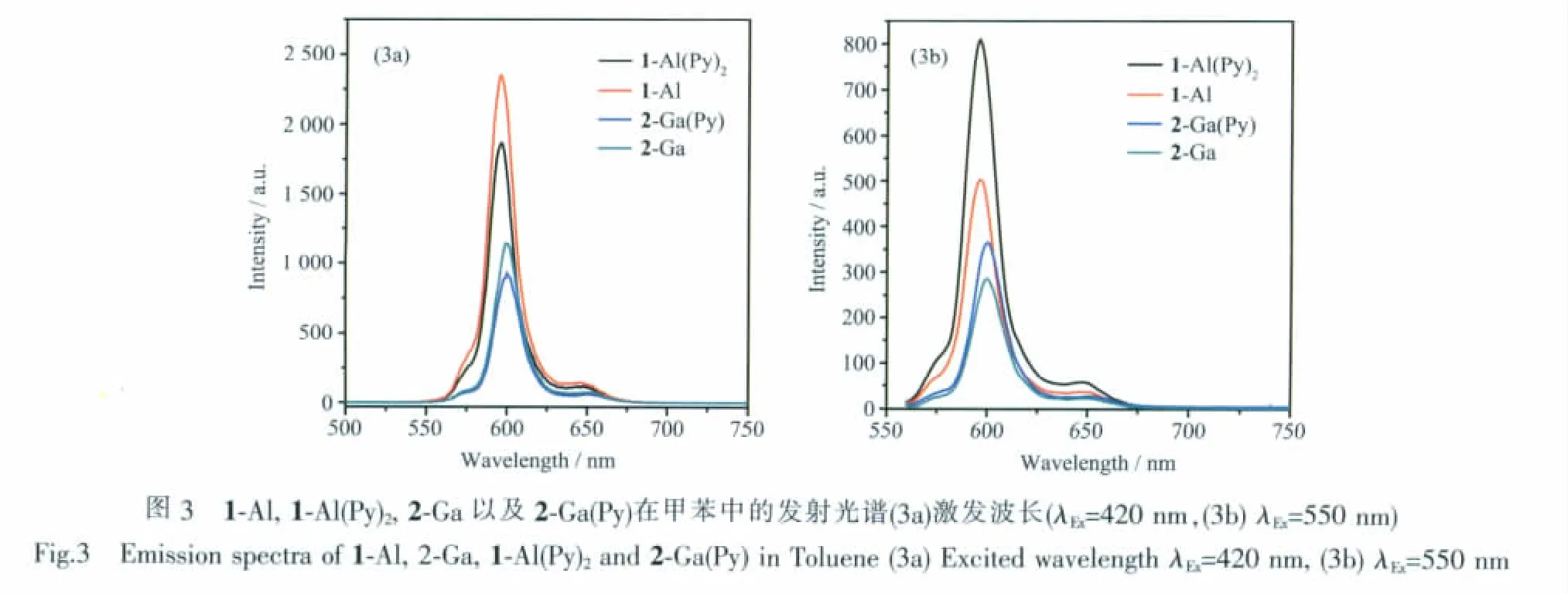
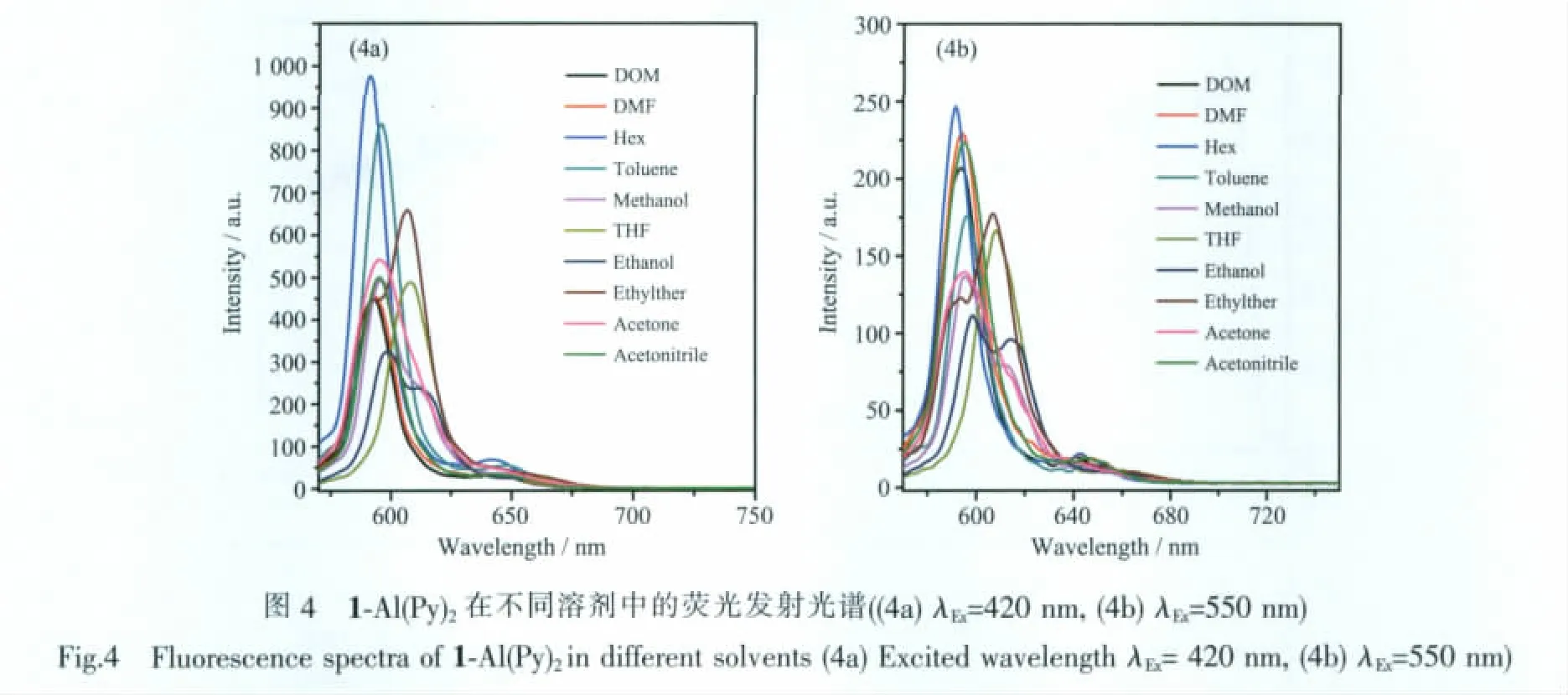
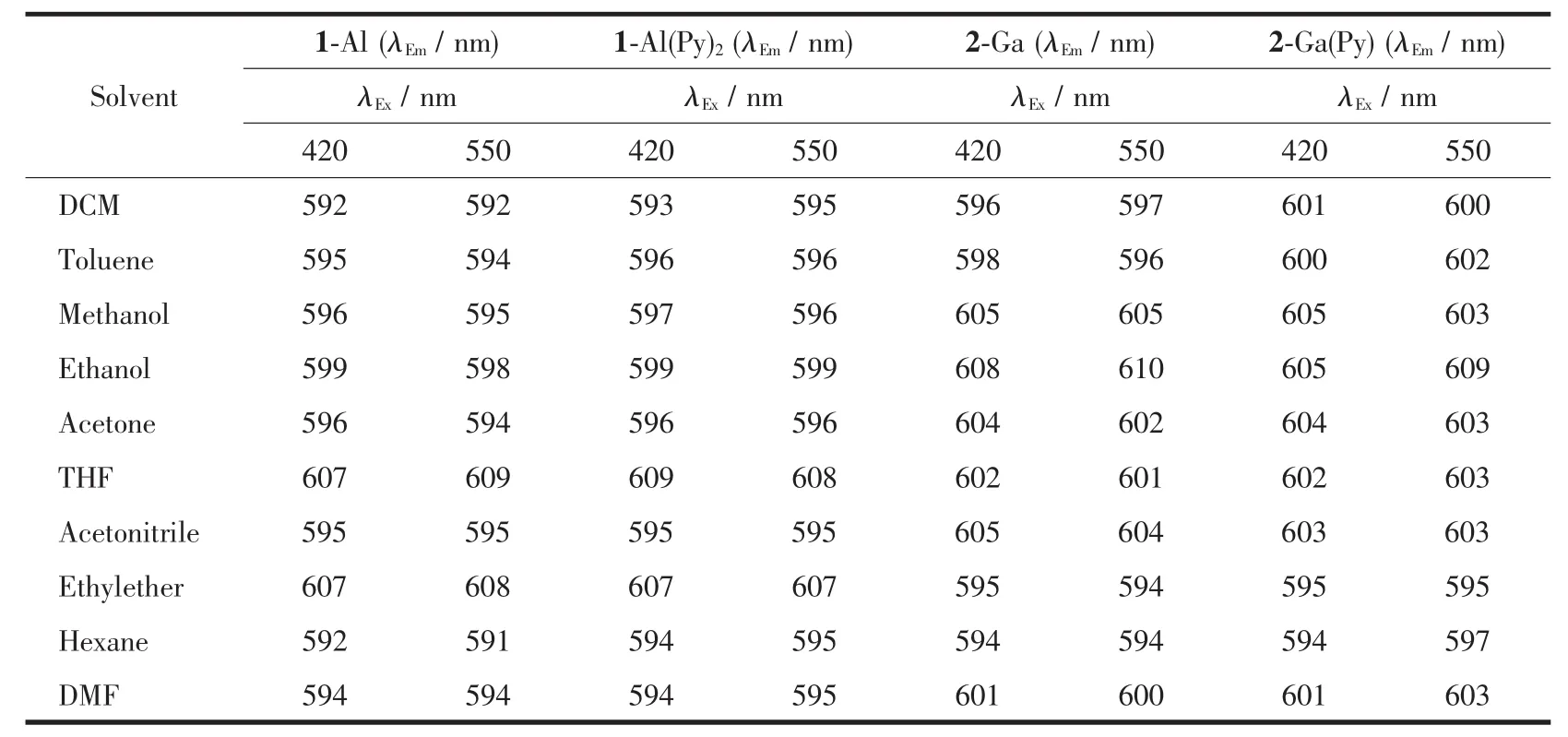
表2 鋁和鎵咔咯配合物在不同溶劑中的發(fā)射光譜數據Table 2 Emission spectra data of aluminum and gallium corroles in different solvents
咔咯和卟啉類化合物具有特殊的光物理性質,平面性好的卟啉類熒光量子產率達0.6[17]左右,卟吩類化合物0.4[18],而鋁和鎵咔咯具有強的熒光性質,1-Al(Py)2和2-Ga(Py)熒光量子產率分別為0.76和0.37[14],在非軸向配位的溶劑甲苯中,2-Ga(Py)的熒光量子產率為0.37,引入軸向配體吡啶的溶劑中,熒光量子產率有所提高,在5%吡啶和甲苯溶液中,1-Al(Py)2和2-Ga(Py)分別為0.76和0.40,當在軸向配位溶劑吡啶中時,2-Ga(Py)達到0.47。以TPP[(φf=0.11)[19]]為標準物,1-Al、1-Al(Py)2、2-Ga、2-Ga(Py)的熒光量子產率分別 0.71,0.74,0.35,0.40, 與部分研究過的結果基本吻合。熒光量子產率的大小與光物理過程熒光發(fā)射過程有關,熒光發(fā)射、內轉換、系間竄越等過程是彼此相互競爭的物理過程,具有重金屬鎵的咔咯配合物,由于重原子效應,其旋軌耦合作用增大,從而增大系間竄越過程,而鋁咔咯的鋁是輕金屬,同時由于其6-配位增大平面剛性,減少了內轉換過程,因而鋁咔咯(0.74)相比鎵咔咯(0.40)具有很大的熒光量子產率。
配合物處于激發(fā)態(tài)時不穩(wěn)定,通過輻射和無輻射躍遷等途徑回到基態(tài),具有銀納米顆粒的卟啉類化合物會影響其Q帶的熒光壽命[20],為了進一步了解中心金屬對咔咯的瞬態(tài)動力學過程的影響,采用條紋相機測定了4種配合物的熒光壽命,通過單指數擬合得到各咔咯配合物的熒光壽命。圖5示4種配合物的熒光衰減曲線,經單指數擬合,得到帶軸向配體吡啶的1-Al(Py)2和2-Ga(Py)的壽命(τ分別為3.66 ns和2.95 ns)較無軸向配體的1-Al和2-Ga(2.85 ns,2.59 ns)長。鎵咔咯的熒光壽命由于重原子效應作用較鋁咔咯短,中心金屬的電子結構和重原子效應會對熒光量子產率和熒光壽命產生一定的影響。同時,結合4種配合物的熒光量子產率和熒光壽命分別得到了其輻射躍遷速率常數kr和無輻射躍遷速率常數knr[6b]。鎵咔咯由于鎵金屬的重原子效應,系間竄越增強,其無輻射躍遷過程較鋁容易進行。而鋁咔咯由于高的熒光量子產率使得其輻射躍遷速率常數明顯大于鎵咔咯配合物的。表3是四種金屬咔咯配合物的發(fā)光動力學參數。不同的中心金屬以及周邊推拉電子取代基對光物理過程的影響在進一步研究中。
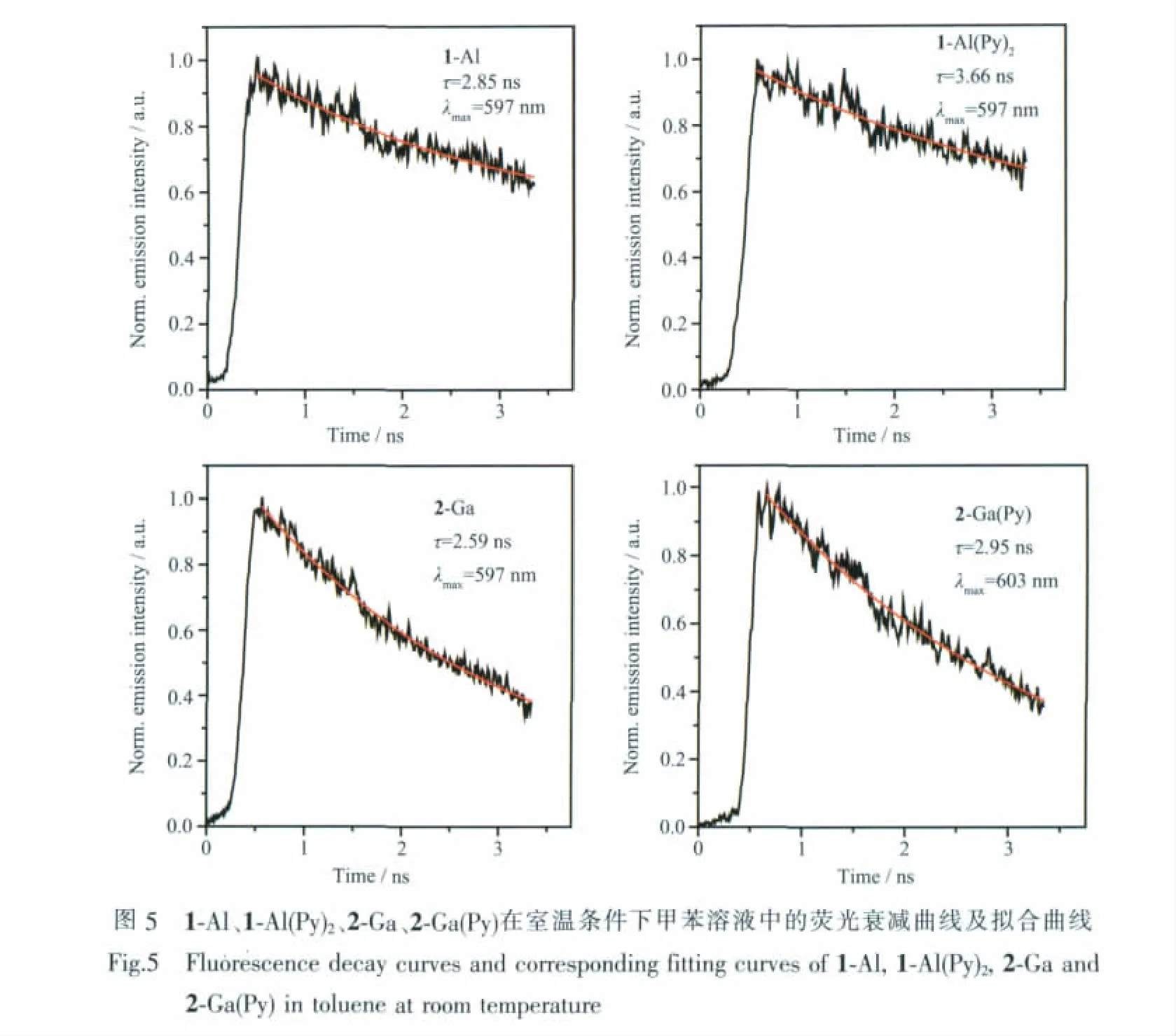

表3 鋁和鎵咔咯配合物的熒光量子產率、熒光壽命、輻射躍遷速率常數和無輻射躍遷速率常數Table 3 Fluorescence quantum yield,fluorescence lifetime,radiative and nonradiative decay rate constants of aluminum and gallium corroles
2.3 π-π 堆積效應
具有π電子共軛體系[21]的卟啉類化合物,在溶液中存在π-π相互作用和金屬-π相互作用[22-23]。Hunter[24]對π-π相互作用建立了理論模型。RHF/STO-3G方法對平行三明治、反平行三明治、平行錯位和平行斜錯位4種幾何構型二聚集的單點能計算結果顯示反平行三明治式結構是最好的堆積方式[25],我們發(fā)現(xiàn)咔咯及其銅配合物在溶液中的π-π堆積作用很弱[26],該結果為硝基Cu咔咯的單晶衍射實驗所證實[27-28]。低溫環(huán)境下,咔咯錳配合物在不同溶劑中因弱的π-π堆積作用而導致不同的磁學性質[29]。第三主族咔咯配合物在溶液中的π-π堆積作用研究尚未見報道,在此,我們利用紫外-可見光譜法[30]考查了所合成第三主族咔咯配合物在二氯甲烷溶液中的π-π堆積作用。
1-Al、2-Ga、1-Al(Py)2和 2-Ga(Py)4 種配合物的最大吸收峰分別在417、418、418、420 nm處,在二氯甲烷溶液中,Soret帶的吸光度會隨濃度增大而增大,當濃度達到一定的數值時,Soret帶就會分裂成2個吸收峰,如圖6所示。4種配合物可能存在π-π堆積效應。
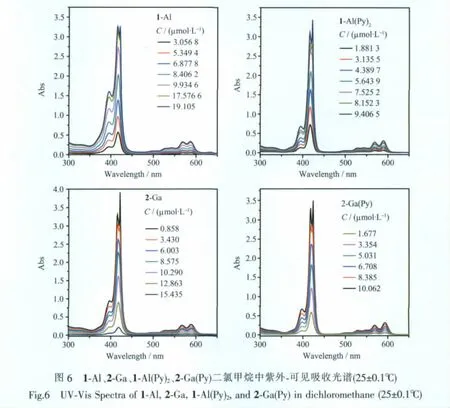

當濃度比較低時,金屬咔咯的吸光度與濃度呈線性關系,當濃度達到一定值后,線性關系發(fā)生明顯改變,拐點的出現(xiàn)說明π-π堆積作用更加明顯。圖7表示了在Soret帶最大吸收處配合物的摩爾消光系數隨濃度的變化情況,在一定濃度范圍內其摩爾消光系數的變化不大,隨著濃度的升高,摩爾消光系數逐漸減小,發(fā)生π-π堆積作用。當4種配合物濃度分別達到 16.048 2 μmol·L-1、15.004 μmol·L-1、6.897 7 μmol·L-1、7.636 9 μmol·L-1時發(fā)生明顯的π-π堆積,帶軸向配體吡啶的 1-Al(Py)2、2-Ga(Py),由于軸向配位的吡啶之間存在具π-π堆積作用,使得 1-Al(Py)2、2-Ga(Py)較 1-Al、2-Ga 更容易發(fā)生聚集。
2.4 軸向配位作用
Corrole與軸向配體間的配位反應可表示為:MC+n L ? MC·n L
n是軸向配體L的化學計量數。在配體L過量的情況下,根據平衡移動原理建立方程[31]:
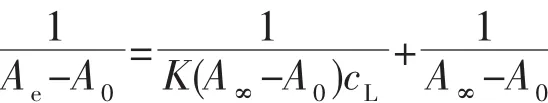
其中,K為平衡常數;A0為不加配體時金屬corrole溶液的吸光度;Ae為加入配體后達到平衡時溶液的吸光度;A∞為加入配體的量為無窮大時溶液的吸光度;CL為配體的濃度(mol·L-1)。1/(Ae-A0)為參變量,1/CL為自變量線性擬合,即可通過斜率a和截距b計算K。
我們選用了咪唑、4-甲基咪唑、吡啶3種配體來考查1-Al和2-Ga的軸向配位作用,隨著軸向配體濃度逐漸增大,Soret帶和Q帶的吸收強度逐漸增大,說明咪唑、4-甲基咪唑、吡啶都能與1-Al和2-Ga相互結合,圖8顯示不同濃度咪唑與2-Ga配位的紫外-可見吸收光譜的變化情況。
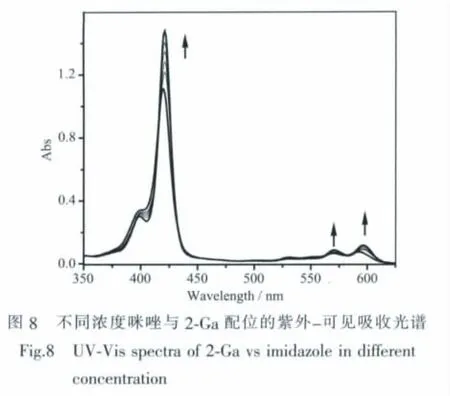
我們分析在421 nm處,體系吸光度值隨咪唑濃度增加的變化情況,以1/(Ae-A0)(Ae,A0分別為平衡時和初始時的吸光度值)為參變量,1/CL(CL為配體濃度 mol·L-1)為自變量進行線性擬合,得到咪唑與 2-Ga 的配位常數為 5.16×105L·mol-1。
如表4中數據所示,咪唑、4-甲基咪唑和吡啶與2-Ga 的配位常數分別為 2.10×105L·mol-1,1.79×105L·mol-1。1-Al與咪唑、4-甲基咪唑和吡啶的配為常數分別為 8.45×105L·mol-1、2.76×105L·mol-1、1.73×105L·mol-1,配位能力為:咪唑>4-甲基咪唑>吡啶。咪唑類的反饋鍵接受能力 (π-accepting)要強于吡啶[32],使得咪唑類結合能力強于吡啶,而由于空間位阻,4-甲基咪唑與1-Al和2-Ga的配位能力要弱于咪唑[32]。1-Al和2-Ga與吡啶配位結合時,其配位數實驗值分別1.84和2.52,即軸向配位的吡啶數目為2。不同于六配位的1-Al(Py)2,合成2-Ga(Py)的時候,合成表征的數據表明,得到的配合物2-Ga(Py)為五配位的。用吡啶滴定的過程中,達到最終的滴定狀態(tài),兩者均為六配位的配合物。而1-Al配合物因以常見的六配位形式存在,配位能力整體強于2-Ga配合物。
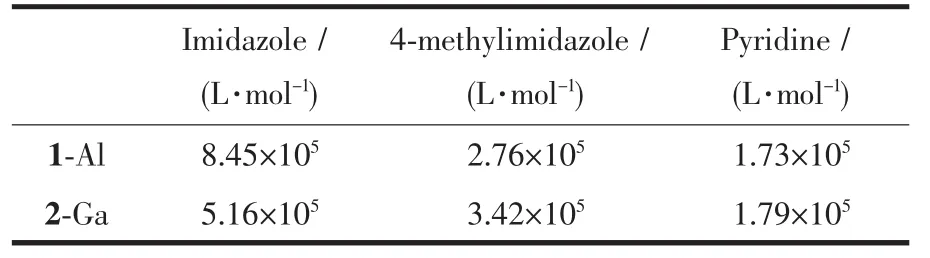
表4 1-Al和2-Ga與咪唑、4-甲基咪唑和吡啶的軸向配位常數Table 4 Axial coordination constant of 1-Al and 2-Ga with imidazole,4-methylimidazole and pyridine
3 結 論
本文制備并確證2種第三主族金屬鋁和鎵的4種金屬咔咯配合物 1-Al、2-Ga、1-Al(Py)2、2-Ga(Py),研究其在不同溶劑中的電子吸收光譜和熒光發(fā)射光譜等光譜性質以及溶液中的自聚集和軸向配位作用。結果表明,配合物最大吸收和發(fā)射峰以及強度因溶劑的介電常數和極性等因素存在差異,鋁和鎵咔咯的熒光壽命和量子產率明顯大于卟啉,在熒光探針和光分子器件方面具有潛在的應用價值。同時,帶軸向配體吡啶的1-Al(Py)2、2-Ga(Py)熒光壽命和量子產率分別大于不帶軸向配體的1-Al、2-Ga,重原子Ga的咔咯配合物壽命相比輕金屬鋁咔咯要短。 1-Al(Py)2、2-Ga(Py)在二氯甲烷溶液中比 1-Al、2-Ga 有利于發(fā)生 π-π 堆積。1-Al、2-Ga 與咪唑、4-甲基咪唑和吡啶相互作用時,軸向配體間結合能力逐漸遞減,而鋁咔咯的結合能力較鎵咔咯大。
[1]Zdilla M J,Abu O,Mahdi M.Inorg.Chem.,2008,47(22):
10718-10722
[2]Flamigni L,Gryko DT.Chem.Soc.Rev.,2009,38:1635-1646[3]Yang Y,Dagmar J,Theodore V H,et al.J.Phys.Chem.,2012,116:1023-1029
[4]Roos B O,Veryazov V,Conradie J,et al.J.Phys.Chem.B,2008,112:14099-14102
[5]Palmer J H,Day M W,Wilson A D,et al.J.Am.Chem.Soc.,2008,130:7786-7787
[6](a)Wagnert L,Berg A,Stavitski E,et al.Appl.Magn.Reson.,2006,30:591-604(b)Liu X,Mahammed A,Tripathy U,et al.Chem.Phys.Lett.,2008,459:113-118(c)Kowalska D,Liu X,Tripathy U,et al.Inorg.Chem.,2009,48:2670-2676
[7](a)Amelia M A,Jeanet C,Peter D W B,et al.J.Am.Chem.Soc.,2008,130:2888-2889(b)Amelia M A,Jeanet C,Ghosh A,et al.Dalton Trans.,2008,37(33):4464-4473
(c)Brothers PJ.Chem.Commun.,2008,44(18):2090-2102
(d)Albrett A M,Peter D W B,George R C,et al.Dalton Trans.,2010,39:4032-4034
(e)Ghosh A,Jynge K.Chim.Eur.J.,1991,3(5):823-833
[8]Agadjanian H,Ma J,Rentsendorj A,et al.PNAS,2009,106(15):6105-6110
[9]Hwang J Y,Lubow J,Chu D,et al.Mol.Pharmaceutics,2011,8:2233-2243
[10]Lim P,Mahammed A,Okun Z,et al.Chem.Res.Toxicol,2012,25:400-409
[11]Mahammed A,Gross Z.Dalton Trans.,2010,39:2998-3000
[12]Vestfrid J,Botoshansky M,Palmer J H,et al.J.Am.Chem.Soc.,2011,133(33):12899-12901
[13](a)Gross Z,Galili N,Simkhovich L,et al.Org.Lett.,1999,1(4):599-602
(b)Gross Z,Galili N,Saltsman I.Angew.Chem.Int.Ed.,1999,38(10):1427-1429
(c)Paolesse R,Jaquinod L,Nurco DJ,et al.Chem.Commun.,1999,14:1307-1308
[14]Mahammed A,Gross Z.J.Inorg.Biochem.,2002,88:305-309
[15]Bendix J,Dmochowski I J,Gray H B.Angew.Chem.Int.Ed.,2000,39(22):4048-4051
[16]Owens J W,Smith R,Robinson R,M,et al.Inorg.Chim.Acta,1998,279:226-231
[17]Braslavsky SE,Muller M,Martire D O,et al.J.Photochem.Photobiol,1997,40:191-198
[18]LIU Wen-Yuan(劉文淵),ZHANG Fu-Shi(張復實),TANG Ying-Wu(唐應武),et al.Acta Phys.-Chim.Sin.(Wuli Huaxue Xuebao),1985,1:12-21
[19](a)Seybold PG,Martin G.J.Mol.Spectrosc.,1969,31:1-13
(b)Dupuis B,Michaut C,Jouanin I,et al.Chem.Phys.Lett.,1999,300:169-176
[20]YANG Zhen-Ling(楊振嶺),LIU Yu-Qiang(劉玉強),YANG Yan-Qiang(楊延強).Acta Phys.Sin.(Wuli Xuebao),2012,61(3):037805-1-037805-5
[21]Hameren R,Elemans JA AW,Wyrosiek D.J.Mater.Chem.,2009,19:66-69
[22]Leighton PCA,Abraham RJ,Sanders JK M.J.Org.Chem.,1988,5:733-740
[23]Hunter CA,Meah M N,Sanders JK M.J.Am.Chem.Soc.,1990,112(15):5773-5780
[24]Hunter C A,Sanders J K M.J.Am.Chem.Soc.,1990,112(14):5525-5534
[25]Chen Y Y,Si L P,Liu J J,et al.Comput.Appl.Chem.,2009,26(12):1587-1592
[26]LIU Hai-Yang(劉海洋),GUO Ping-Ye(郭平葉),XU Zhi-Guang(徐志廣),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2007,23(3):504-508
[27]Dibyendu B,Pinky S,Sabyasachi S.Inorg.Chim.Acta,2010,363:4313-4318
[28]Miao X R,Gao A M,Li Z M,et al.Appl.Surf.Sci.,2009,255:5885-5890
[29]Ding M,Wang B W,Wang Z M,et al.Chem.Eur.J.,2012,18(3):915-924
[30]WU Di(吳迪),WU Jian(吳健).Chem.World(Huaxue Shijie),2004,10:551-555
[31]Miller J R,Dorough C D.J.Am.Chem.Soc.,1952,74(16):3977-3981
[32]Yamamoto K.Inorg.Chim.Acta,1986,113:181-186
