不同形貌ZnO@PANI納米復合材料的制備及光催化性質
2013-09-15 03:03:32吳振玉李奉杰朱維菊
無機化學學報 2013年10期
關鍵詞:復合材料
吳振玉 李奉杰 李 村 朱維菊 方 敏
(安徽大學化學化工學院,合肥 230601)
0 引 言
近年來,隨著環境污染的加劇,控制污染、保護環境,實現可持續發展是人們的共同愿望。半導體光催化成為污染控制化學研究的一個熱點,是一種具有廣闊應用背景的綠色環境治理技術,其中光催化降解是指半導體光催化劑在光照條件下可以產生具有強氧化性的OH·自由基氧化分解各種有機污染物。一些具有光催化活性的半導體氧化物,如TiO2、ZnO等[1-2],成為研究最多應用最廣的催化材料。納米ZnO由于較低的成本,簡便的合成方法,且與TiO2具有類似的帶隙結構而備受關注[3-11]。其中一些研究結果表明,棒狀納米ZnO甚至表現出比TiO2更高的光催化效率[1-2];Xie等也發現制備方法和形態對納米ZnO的光催化性能影響較大[12];Gou等發現ZnO納米棒陣列展現出優越的光催化降解有機染料的能力,花狀納米ZnO次之,顆粒狀納米ZnO相比較差[13]。這些結果說明納米ZnO的形貌、尺寸和納米結構等因素都會影響半導體的光催化效率。
納米ZnO作為一種常用的半導體光催化劑使用中還存在一些不足,如易于受到光腐蝕,化學穩定性不高。此外純納米ZnO對太陽光的主要吸收波長范圍在紫外區,且光生載流子的復合率較高,其光催化效率不是很高。所以提高ZnO光催化劑的穩定性和光催化活性成為研究熱點。目前常用的方法就是對ZnO半導體光催化劑進行摻雜或復合[7-8,11,14-16],一般認為摻雜或復合有利于形成更多的氧空穴[14-15],或者可以加快光生電子和空穴的分離,抑制載流子復合[11,16],從而提高光催化材料的光催化效率。
聚苯胺具有特殊的質子摻雜性、良好的氧化還原性和環境穩定性以及較高的摻雜電導率,易于大量生產和成本相對低廉等優點[3]。將制備成本相對較低的半導體材料納米ZnO與制備成本相對低廉的聚苯胺復合,可以提高ZnO的光腐蝕保護能力及穩定性[17-18],改善ZnO的電學性質、介電性能、光學性質等[19-25]。
在ZnO-PANI復合材料的研究中,光催化性質的研究相對較少,而且已有的研究中,還存在一定的不足。現在已經報道的ZnO/PANI復合物一般是采用與PANI共混[17]或者直接采用苯胺在ZnO表面聚合[3,18,26-27],可能由于聚合物與納米ZnO的界面之間結合作用不強,已報道的ZnO-PANI復合材料的催化效率不是很高。為改善PANI/ZnO復合材料的光催化性能,在課題組前期納米ZnO制備研究的基礎上[28],通過簡單易控的方法合成了幾種不同形貌的納米ZnO,使用KH-42以回流法對其表面進行改性,然后經由皮克林乳液聚合法,制備出納米ZnO-聚苯胺復合材料,提高了納米ZnO的光催化降解性能,得到了在紫外光和可見光中均有良好催化效率的納米復合材料光催化劑。
1 實驗部分
1.1 試劑與儀器
二水合醋酸鋅(分析純,國藥試劑),氯化鋅(分析純,國藥試劑),氫氧化鈉(分析純,國藥試劑),無水乙醇(分析純,國藥試劑),超純凈水(艾科浦純水器制備,重慶頤洋)。硅烷偶聯劑KH-42(分析純,江蘇金壇市硅烷偶聯劑廠),過硫酸銨(分析純,國藥試劑),二甲苯(分析純,國藥試劑),甲苯(分析純,國藥試劑),苯胺(分析純,國藥試劑),醋酸(分析純,國藥試劑),亞甲基藍(MB)(分析純,國藥試劑)。
XD-3型X射線衍射儀(北京普析通用儀器有限責任公司),NEXUS-870型傅里葉紅外光譜儀 (美國Nicolet Instrument Corporation公司)。 Lamda-900型紫外-可見-近紅外光譜儀(美國Perkin-Elmar公司),F-4500熒光光譜儀 (日本日立公司),S-4800型掃描式電子顯微鏡 (日本日立公司),JEM-2100高分辨透射電子顯微鏡(日本電子公司)。449F3型同步熱分析儀(德國耐馳公司)。PLS-SXE300/300UV 型氙燈(北京暢拓科技公司)。
1.2 實驗過程
1.2.1 納米ZnO的合成
直接液相沉淀法溫控合成顆粒狀和棒狀納米ZnO[28]: 稱 取 4.42 g 的 ZnAc2·2H2O 和 1.60 g 的NaOH,分別溶解在100 mL無水乙醇溶劑中,磁力攪拌下,將NaOH溶液快速倒入到裝有ZnAc2·2H2O溶液的圓底燒瓶中,反應圓底燒瓶上加回流冷凝管,分別在25、80℃下恒溫攪拌24 h,離心分離,依次用無水乙醇、去離子水洗滌數次,室溫真空干燥,分別得到顆粒狀和棒狀納米ZnO。
水熱法控制合成球形花狀納米ZnO:稱取1.36 g ZnCl2,溶解在10 mL去離子水中,加入事先配置好的1 mol·L-1NaOH溶液20 mL,充分混合。將所得的混合液加入帶有50 mL聚四氟乙烯內襯的反應釜中,再加入5 mL左右的去離子水120℃水熱反應12 h,自然冷卻至室溫,將反應釜內的產物抽濾,依次用去離子水和無水乙醇各洗滌3次,80℃真空干燥12 h,得到球形花狀納米ZnO。
1.2.2 ZnO@PANI納米復合材料的制備
納米ZnO的表面化學修飾:將0.5 g納米ZnO加入圓底燒瓶中,然后加入0.1 mL的KH-42、10 mL二甲苯、0.5 mL H2O,攪拌均勻,80 ℃回流 3 h,離心分離,依次用去離子水和無水乙醇各洗滌3次,80℃真空干燥12 h,得到m-ZnO樣品。
HAc摻雜室溫皮克林乳液法制備納米m-ZnO@PANI復合材料:m-ZnO@PANI復合材料的制備參照文獻[29],具體過程如下,將0.4 g過硫酸銨(APS)溶解在 50 mL水中,加入 2 mL 10%HAc,攪拌均勻,得到溶液A。在裝有6 mL甲苯的圓底燒瓶中加入 0.24 mL單體苯胺,攪拌均勻后,加入0.4 g m-ZnO,得到乳液B。室溫下將A溶液滴加到乳液B中,室溫反應3 h,抽濾,依次用去離子水和無水乙醇各洗滌 3次,60℃真空干燥 12 h,得到 m-ZnO@PANI納米復合材料。
1.2.3 材料的光催化性能實驗
在100 mL濃度為15 mg·L-1的亞甲基藍溶液中,加入30 mg復合材料催化劑樣品,避光黑暗攪拌30 min,然后在300 W氙燈照射下進行光催化降解,每隔一段時間取一次樣,離心分離,測亞甲基藍溶液在665 nm處的吸光度,根據標準工作曲線計算出相應的亞甲基藍濃度。光催化光源分別使用組合濾光片選擇320~780 nm的紫外-可見光和400~780 nm的可見光,所有光催化反應均在室溫下進行。
脫色率按照下面的公式計算:脫色率D=(c0-c)/c0×100%。c0為亞甲基藍溶液的初始濃度,c為光照一段時間后取出樣品離心所得上清液的濃度。
2 結果與討論
2.1 材料的表征
2.1.1 納米 ZnO 的表征
圖1為不同制備條件下所得樣品的XRD圖,圖中所有樣品的衍射峰都可以清楚地指標化為純的六方相纖鋅礦結構的ZnO晶面 (PDF No.36-1451)。XRD圖中沒有出現其它雜質的衍射峰,表明在這些條件下制得的產物為純凈的ZnO。在25℃直接沉淀法制備的樣品衍射峰強度較低,峰形有些寬化,表明室溫下ZnO納米晶粒尺寸較小,隨著反應溫度的升高,樣品的衍射峰強度逐漸增大,峰形進一步銳化,顯示生成ZnO納米晶粒度變大、結晶度增加。

圖1 不同制備條件下所得納米ZnO的XRD圖Fig.1 XRD patterns of ZnO nanomaterials prepared at different condition
圖2 a和2b所示為直接沉淀法制備納米ZnO時,在不同反應溫度下所得ZnO的SEM圖。從圖中我們可以看到,通過簡單地調節反應溫度即可控制產物的形貌。室溫下(25℃)得到尺寸較均勻和分散性較好顆粒狀納米ZnO(圖2a),粒徑約為30 nm;80℃時,ZnO的形貌已經與25℃時有很大的不同,為分散性和大小都比較均勻的ZnO納米棒 (圖2b),納米棒的長度100 nm左右,直徑約40 nm。這表明,溫度對ZnO棒狀結構的生長起到了重要的作用,文獻指出氧化鋅各向異性的生長特性只在一定的溫度下才能明顯地表現出來[28]。
圖2c和2d是120℃水熱法時所得樣品的SEM圖,從圖中能夠看出,產物為由片層狀結構聚集的球形花狀ZnO,分散性和大小都比較均勻,片層厚度約20 nm,球體直徑約2.0μm。
以上分析結果說明可以通過反應溫度和制備方法比較容易地控制納米ZnO的形貌,采用直接沉淀法制備的時候,當反應溫度為25℃時,所得產品呈分散度和大小都比較均勻的零維顆粒狀納米ZnO,80℃時為分散度和大小都比較均勻的一維棒狀納米ZnO。而采用水熱反應法制備的時候,水熱反應溫度為120℃時,得到分散度和大小都比較均勻的球形花狀ZnO,從而實現了納米ZnO的簡單易控合成。
2.1.2 m-ZnO@PANI復合材料的表征

圖2 不同條件下所得納米ZnO的SEM圖Fig.2 SEM images of ZnO prepared at different condition
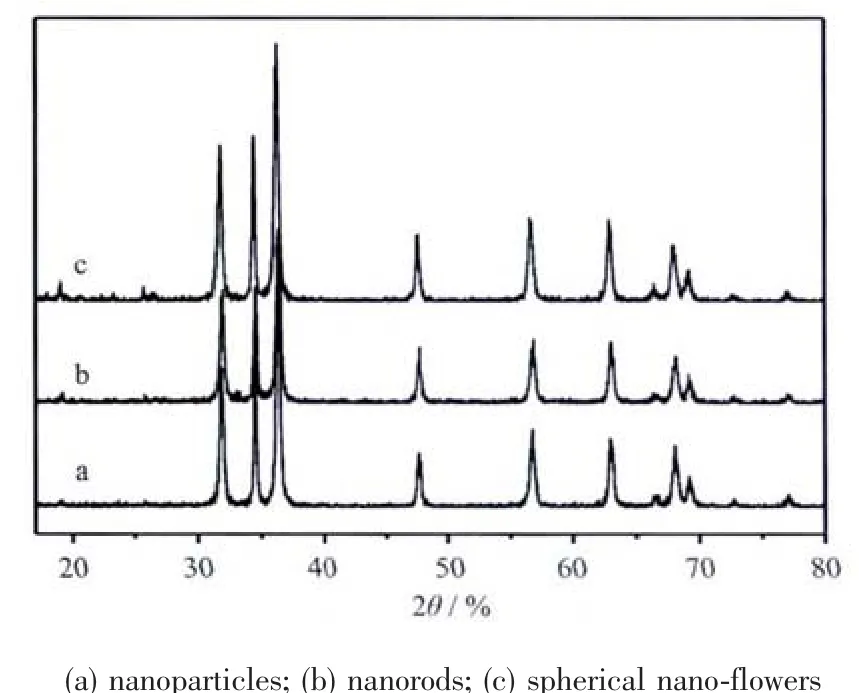
圖3 不同形貌m-ZnO@PANI復合材料的XRD圖Fig.3 XRD patterns of the different morphological m-ZnO@PANI nanocomposite
圖3 是不同形貌的改性納米ZnO,與苯胺單體通過室溫皮克林乳液法制得的復合材料的XRD圖,所有樣品的較強衍射峰均可以指標化為六方相纖鋅礦結構ZnO晶體的相應晶面,說明化學改性以及乳液聚合過程沒有改變納米ZnO的晶體結構,在氧化鋅表面形成的聚苯胺基本上以非晶態存在。此外,圖3b、3c中在2θ為19°和26°左右出現了弱的衍射峰,這可能是由于聚苯胺在ZnO片狀納米花瓣表面聚合時,形成局部有序的晶相微區,表現了一定程度的低結晶度,其中26°左右的峰可能為PANI鏈在空間重復所形成的低度結晶[18],而19°左右的峰可能為低度結晶的PANI鏈中苯環和醌環的重復[18,20]。同樣的實驗條件,圖3a的PANI衍射峰幾乎沒有,圖3b的PANI衍射峰也不如圖3c的強,說明納米ZnO形態和表面大小可能影響PANI的結晶情況,通常棒狀的ZnO-PANI復合物中PANI的結晶度[18]好于顆粒狀的ZnO-PANI[20,27]。從圖3的結果來看,可能球形花狀ZnO的平整片層結構更有利于PANI的結晶。
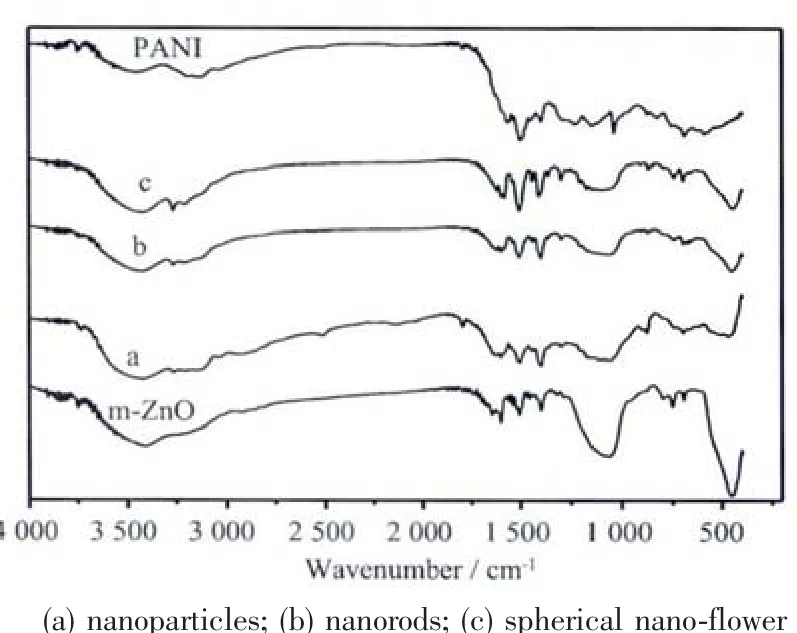
圖4 不同形貌m-ZnO@PANI復合材料的FTIR譜圖Fig.4 FTIR spectra of the different morphological m-ZnO@PANI nanocomposite
從圖4可以看到,改性后在2 900 cm-1附近是CH2-的特征吸收峰,在1 600 cm-1處出現了-NH特征吸收峰,1 500 cm-1,500 cm-1處出現了苯環特征峰,1 100 cm-1為Si-O-M(M=Si或Zn)特征吸收峰,表明硅烷偶聯劑KH-42已經通過化學作用接枝在納米ZnO表面。m-ZnO@PANI復合材料的FTIR圖譜和m-ZnO的相同之處是,均在1 100 cm-1附近出現了較寬的Si-O-M伸縮振動峰。此外,在1 450~1 650 cm-1之間出現了尖銳、較多的PANI特征峰,其中1 504 cm-1處為醌環的伸縮振動特征峰,1 410 cm-1處為苯環的伸縮振動峰,表明苯胺單體被氧化為聚苯胺[3,18]。
圖5為所制備材料的UV-Vis譜圖,從圖中可以看到,KH-42改性的m-ZnO納米球與純納米氧化鋅的吸收光譜基本相似,在紫外區出現了很強的吸收峰;m-ZnO@PANI和m-ZnO以及PANI相比,不僅在紫外區出現了較強的寬帶吸收,而且在可見光區也有比較強的吸收,結果表明氧化鋅聚苯胺的復合可以拓展對太陽光中可見光的吸收和利用。

圖5 不同樣品的UV-Vis譜圖Fig.5 UV-Vis spectra of different samples
圖6 是不同形貌m-ZnO與苯胺單體通過皮克林乳液聚合所得的m-ZnO@PANI復合材料的高分辨透射電鏡圖。從圖中看到,有機聚苯胺包覆在ZnO表面,形成緊密的異質界面,其包覆層大約1~2 nm,這是由于ZnO表面化學修飾的硅烷偶聯劑上苯胺基團可能與溶液中苯胺分子發生氧化偶聯,形成的聚苯胺分子鏈直接錨接在納米氧化鋅表面,由于球形花狀的特殊結構,其表面包覆的PANI相對較多,樣品的熱重分析結果也證實這點。
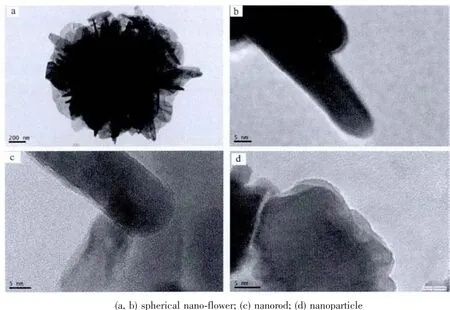
圖6 不同形貌m-ZnO@PANI復合材料的TEM圖Fig.6 TEM images of different morphological m-ZnO@PANI nanocomposites
圖7 為改性后ZnO通過室溫皮克林乳液法與苯胺單體聚合所得復合材料的TG圖,可以看到,m-ZnO在220℃左右受熱失水約3%,與聚苯胺復合后,和m-ZnO相比由于聚苯胺受熱分解,失重明顯增多,隨著反應時間延長,失重率先增大后減小,而且花狀納米ZnO復合物失重最多,棒狀次之,顆粒狀最少。失重情況與高分辨電鏡觀察復合材料中所含PANI的多少一致。
2.2 材料的光催化性能
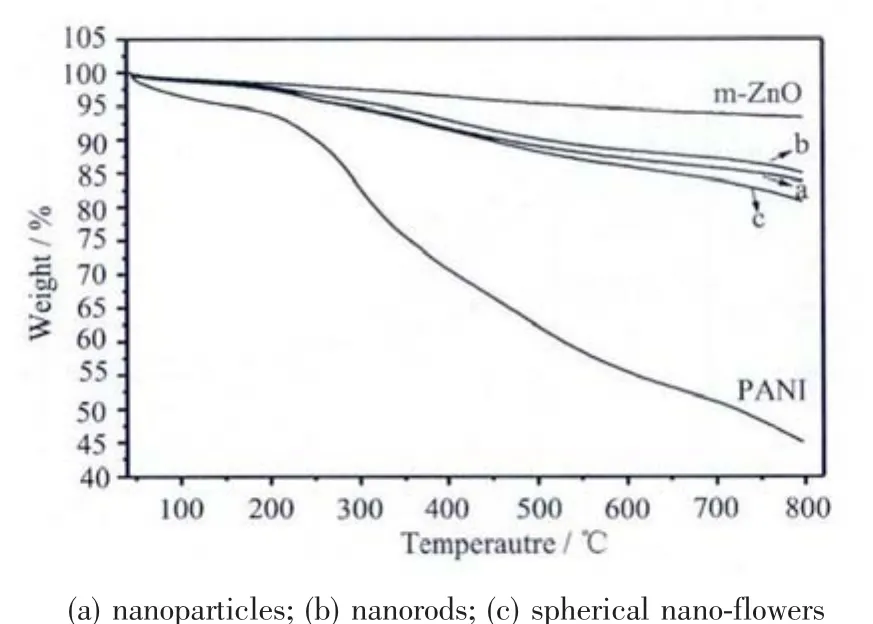
圖7 m-ZnO@PANI的TG圖Fig.7 TGcurves of m-ZnO@PANI
圖8 是不同形貌納米ZnO與PANI的復合材料在紫外-可見光(a)和可見光(b)催化下對亞甲基藍的脫色率圖。從圖8a看出,不同形貌的納米復合材料都對亞甲基藍光降解反應有催化作用,其中ZnO納米棒和納米花復合材料在紫外光照射下具有很高光催化性能,光催化40 min后脫色率均超過90%,最高可達98.2%;ZnO納米顆粒形成的復合材料光催化活性較低,光催化70 min后脫色率可達到80%。從圖8b可以看到,在可見光范圍內,m-ZnO與聚苯胺的納米復合材料光催化活性明顯降低,光催化反應4 h后,亞甲基藍脫色率才超過50%,其中納米棒形成的復合材料光催化效率最大,脫色率可超過95%,納米花形成的復合材料脫色率超過70%,延長光照時間在5 h以上,脫色率可以達到跟紫外-可見光的效果相近。文獻報道也說明純PANI[26]和純納米ZnO[27]在的可見光光催化能力都很弱,當它們形成復合材料以后,具有可見光響應的光催化性能。雖然可見光催化降解率也比較大,但是達到高降解率光照的時間遠遠大于紫外可見光。

圖8 不同形貌m-ZnO@PANI納米復合材料在紫外-可見光(a)和可見光(b)催化下對亞甲基藍的脫色率Fig.8 photodegradation efficiency on MB of m-ZnO@PANI nanocomposites irradiation by UV-Vis(a)and Visible(b)light
催化劑在光催化過程中受到諸多因素的影響,如尺寸大小、結晶形態、晶體結構和表面缺陷結構等。納米ZnO的某些晶面上具有更多的缺陷和活性點,與具有許多片狀結構的球狀ZnO和極性生長而成的棒狀ZnO相比,顆粒狀ZnO活性點相對較少,從而導致催化效率相對較低,這與文獻報道的不同形貌純納米ZnO材料光催化結果一致[13]。
我們還發現光催化反應后的復合材料XRD衍射花樣和其紫外-可見吸收光譜也幾乎沒有變化,說明復合材料具有較好的穩定性,可以分離出來而循環使用。為了解納米復合材料的重復使用的光催化性能,將光催化反應結束的球形納米花復合材料光催化劑離心分離后,再加入同樣量的亞甲基藍溶液,按照同樣操作進行二次循環光催化實驗,實驗結果見圖9。從圖中可以看到,復合材料表現出很好的循環光催化活性和效率,第二次循環光催化反應60 min的脫色率仍達到了96%。表明所制備的納米ZnO具有很高的穩定性,可以循環使用。
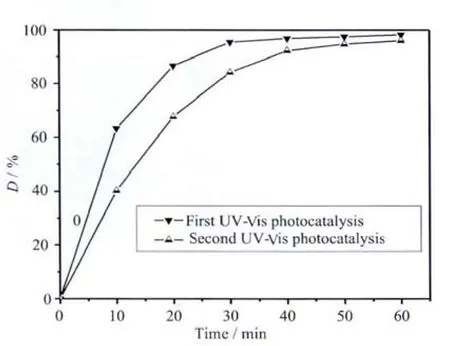
圖9 球形花狀m-ZnO@PANI循環紫外-可見光催化脫色率曲線Fig.9 UV-Vis photodegradation efficiency of spherical flowers-like m-ZnO@PANI nanocomposite
2.3 復合材料制備和光催化機理探討
通過硅烷偶聯劑KH-42對納米ZnO的表面改性,提高它與聚合物的相互作用,增強其穩定性和表面活性。在水的作用下,烷氧基水解為硅醇,然后硅醇羥基與ZnO表面的羥基發生縮合反應,接枝的分子含有苯胺基團,它可以作為聚苯胺分子鏈形成的生長點。在皮克林乳液聚合中,ZnO表面的苯胺基團與分散在乳液中的苯胺分子在過硫酸銨(APS)的作用下發生氧化聚合成聚苯胺包裹ZnO納米顆粒形成ZnO-PANI復合材料。
在ZnO納米材料的表面通過化學修飾,原位聚合,可以形成良好的異質結界面,有利于光生電子-空穴對的分離[30],從而可以提高復合材料的光催化活性和效率,同時利用聚苯胺對可見光、紅外光吸收,可以拓展ZnO光催化劑的太陽光吸收范圍,增強對太陽光中可見光的吸收利用,提高光催化性能。我們制備的復合材料對紫外光和可見光均表現出良好的光催化性能,其在紫外和可見光照條件下的催化過程可以認為:在紫外光照射下,ZnO產生電子-空穴對,電子轉移至ZnO表面發生氧化還原反應,空穴轉移至ZnO與聚苯胺之間界面的最高已占軌道,與H2O結合,生成具有強氧化性的OH·;在可見光照射下,包覆在ZnO表面的聚苯胺吸收可見光,激發電子到最低未占據軌道,產生游離電子e-,e-轉移至復合材料表面發生還原反應,也可生成具有強氧化性的 O2·、OH·[17,26]。 綜上所述,氧化鋅與聚苯胺復合后,可以提高了對太陽光的吸收利用率,有利光照產生e--h+對的分離,在可見光或紫外光照射下都能產生了具有強氧化性的 O2·,HO2·,OH·等自由基,從而高效率催化降解亞甲基藍。
3 結 論
采用直接沉淀法和水熱法,分別制備出形貌和尺寸比較均一的顆粒狀、棒狀和球形花狀的氧化鋅納米材料,使用具有苯胺基團的硅烷偶聯劑對所得納米ZnO進行表面化學改性,再通過皮克林乳液聚合制備了氧化鋅與聚苯胺的復合材料。發現納米復合材料中的ZnO與聚苯胺之間通過化學鍵合形成異質結界面,提高復合材料光催化劑的穩定性以及光催化活性和效率。通過聚苯胺的復合拓展了納米氧化鋅對可見光的吸收,使納米復合材料對紫外光、可見光都有較強吸收,具有很好的紫外光和可見光光催化性能。研究還發現棒狀和花狀納米復合材料的光催化性能比顆粒狀納米復合材料的性能好,光催化降解亞甲基藍的脫色率超過95%;復合材料還有很好光催化穩定性,重復使用時可保持良好的光催化性能,二次循環光催化降解率仍可達到96.0%。
[1]Guo M Y,Fung M K,Fang F,et al.J.Alloys Compd.2011,509:1328-1332
[2]Faisal M,Khan S B,Rahman M M,et al.Appl.Surf.Sci.,2011,258:672-677
[3]SONG Ji-Zhong(宋繼中),HE Ying(賀英),PAN Zhao-Dong(潘照東),et al.Acta Chim.Sin.(Huaxue Xuebao),2011,69:1582-1588
[4]WANG Hu(王 虎 ),XIE Juan(謝 娟 ),DUAN Ming(段 明).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27:193-198
[5]WANG Hu(王 虎 ),XIE Juan(謝 娟 ),DUAN Ming(段 明).Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2011,27:321-326
[6]WANG Zhi-Fang(王志芳),LI Mi(李密),ZHANG Hong-Xia(張紅霞),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2012,28:715-720
[7]GAO Hai-Xia(高海霞),CHENG Guo-Feng(程國峰),CHENG Rong-Ming(成榮明),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2006,22:872-876
[8]LI Yun-Jun(李躍軍),YIN Zhong-Hong(尹忠紅),CAO Tie-Ping(曹鐵平),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebaoe),2011,27:1348-1352
[9]TENG Hong-Hui(滕洪輝),XU Shu-Kun(徐淑坤),WANG Meng(王猛).J.Inorg.Mater.Sin.(Wuji Cailiao Xuebao),2010,25:1034-1040
[10]LI Chang-Quan(李長全),LUO Lai-Tao(羅來濤),XIONG Guang-Wei(熊光偉).Chin.J.Catal.(Cuihua Xuebao).2009,30:1058-1062
[11]YU Chang-Lin(余長林),YANG Kai(楊凱),YU Ji-Mei(余濟美),et al.Acta Phys.-Chim.Sin.(Wuli Huaxue Xuebao),2011,27,505-512
[12]Wang H H,Xie C S,Zhang W,et al.J.Hazard.Mater.2007,141:645-652
[13]Ma SS,Li R,LüCP,et al.J.Hazard.Mater.2011,192:730-740
[14]Mahmood MA,Baruah S,Dutta J.Mater.Chem.Phys.,2011,130:531-535
[15]Patil A B,Patil K R,Pardeshi SK.J.Hazard.Mater.2010,183:315-323
[16]Fu D Y,Han G Y,Chang Y Z,et al.Mater.Chem.Phys.2012,132:673-681
[17]Zhang H,Zong R L,Zhu Y F.J.Phys.Chem.C,2009,113,4605-4611
[18]Eskizeybek V,Sar F,Gülce H,et al.Appl.Cataly.B:Environ,2012,119-120:197-206
[19]Li Y H,Gong J,McCune M,et al.Synth.Metal,2010,160:499-503
[20]Sharma B K,Kharea N,Dhawan SK,et al.J.Alloy.Compd,2009,477:370-373
[21]Huang G W,Xiao H M,Shi H Q,et al.J.Poly.Sci.Part A:Poly.Chem,2012,50:2794-2801
[22]Alves K G B,Felix J F,Melo E F D,et al.J.Appl.Poly.Sci.,2012,125:E141-E147
[23]Sharma S P,Suryanarayana M V S,Nigam A K,et al.Cataly.Comm.,2009,10:905-912
[24]Huang J,Yang T L,Kang Y F.et al.Natural.Gas.Chem.2011,20:515-519
[25]Ahmed F,Kumar S,Arshi N.et al.Thin Solid Films,2011,519:8375-8378
[26]Ameen S,Akhtar M S,Kim Y S,et al.Colloid.Polym.Sci.,2011,289:415-421
[27]Olad A,Nosrati R.Res.Chem.Intermed.,2012,38:323-326
[28]LI Feng-Jie(李奉杰),LI Cun(李村),ZHANG Xian-Li(張現利),etal.Bull.Chin.Ceram.Soc.(Guisuanyan Tongbao),2012,31:145-149
[29]He Y J.Appl.Surf.Sci.,2005,249:1-6
[30]Zhu S,Wei W,Chen X,et al.J Solid State Chem.,2012,190:174-179
猜你喜歡
建材發展導向(2022年2期)2022-03-08 01:44:04
建材發展導向(2021年14期)2021-08-23 00:56:16
中國材料進展(2019年10期)2019-12-07 05:32:14
纖維復合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
山東工業技術(2016年15期)2016-12-01 05:31:34
中國塑料(2015年6期)2015-11-13 03:02:54
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年8期)2015-10-14 01:10:41
應用化工(2014年10期)2014-08-16 13:11:29
