三高調和丸質量標準研究
2013-09-25 11:00:36劉文龍黃余燕
中國現代藥物應用 2013年13期
劉文龍 黃余燕
三高調和丸由丹參、三七、天花粉、澤瀉、甘草等組成,其功效為活血消瘀、消食化積、清熱生津、扶正驅邪。適用于高血壓、高血脂、糖尿病、及脈管炎等癥。此次試驗的目的是完善三高調和丸質量標準,薄層色譜法(TCL)對處方中的丹參、三七這兩種主要藥材進行定性鑒別,并采用用高效液相色譜法(HPLC)測定丹參含量。
1 儀器與試藥
1.1 儀器 Agilent1100高效液相色譜儀。
1.2 試藥 硅膠G由青島海洋化工有限公司提供批號:080222丹參酮ⅡA對照品(批號:110766-200518,中國藥品生物制品檢驗所)。甲醇為色譜純,水為超純水,其他試劑均為分析純。三高調和丸的制備:三高調和丸為嘉應學院醫學院制劑室生產(生產批號:20081118;20081125;20081129)
2 方法與結果
2.1 薄層鑒別
2.1.1 丹參的鑒別
2.1.1.1 供試品溶液的制備 取本品研細,稱取細粉10 g,加乙醚30 ml,超聲處理10 min,振搖,放置1 h,濾過,濾液揮干,殘渣加乙酸乙酯1 ml使溶解,作為供試品溶液。
2.1.1.2 對照品溶液的制備 另取丹參對照藥材1 g,同法制成對照藥材溶液。再取丹參酮ⅡA對照品,加乙酸乙酯制成2 mg/ml的溶液,作為對照品溶液。
2.1.1.3 陰性對照溶液的制備:取相當于樣品量10 g的缺丹參陰性對照樣品,按上述供試品溶液制備方法制備丹參陰性對照溶液。
2.1.1.4 薄層色譜分析 照薄層色譜法(《中國藥典》(2010年版一部附錄VI B)試驗[1],吸取上述三種溶液各10 μl,分別點于同一硅膠G薄層板上,以苯-乙酸乙酯(19:1)為展開劑,展開,取出,晾干。供試品色譜中,在與對照藥材色譜相應的位置上,顯相同顏色的斑點,在與對照品色譜相應的位置上顯相同的暗紅色斑點,并且丹參陰性對照未出現斑點。結果見圖1。
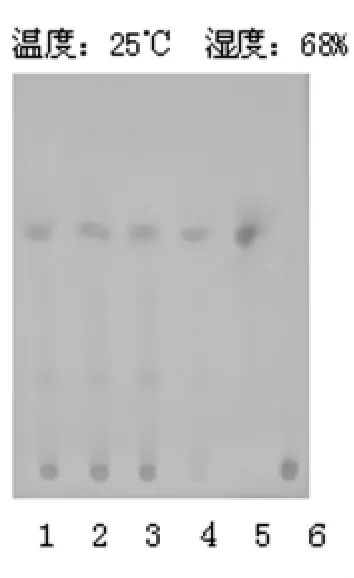
圖1
2.1.2 三七的鑒別
2.1.2.1 供試品溶液的制備 取本品研細,稱取細粉10 g,加水10 ml使其潤濕,攪勻,再加水飽和的正丁醇30 ml,超聲處理30 min,濾過,濾液用正丁醇飽和的水洗滌3次,每次10 ml,棄去水層,取正丁醇提取液,蒸干,殘渣加甲醇1 ml使溶解,作為供試品溶液
2.1.2.2 對照品溶液的制備 另取三七對照藥材0.5 g,同法制成對照藥材溶液。再取三七皂苷R1對照品及人參皂苷Rb1、人參皂苷Rg1對照品,加甲醇制成每1 ml各含1 mg的混合溶液,作為對照品溶液。
2.1.2.3 陰性對照溶液的制備 取相當于樣品量10 g的缺三七陰性對照樣品,按上述供試品溶液制備方法制備三七陰性對照溶液。
2.1.2.4 薄層色譜分析 照薄層色譜法(《中國藥典》(2010年版一部附錄VI B)試驗[1],吸取上述三種溶液各2 μl,分別點于同一硅膠G薄層板上,以三氯甲烷-乙酸乙酯-甲醇-水(15∶40∶22∶10)10℃以下放置的下層溶液為展開劑,展開,取出,晾干,噴以硫酸乙醇液(1→10),在105℃加熱至斑點顯色清晰。供試品色譜中,在與對照藥材和對照品色譜相應的位置上,顯相同顏色的斑點,并且三七陰性對照未出現斑點。結果見圖2。
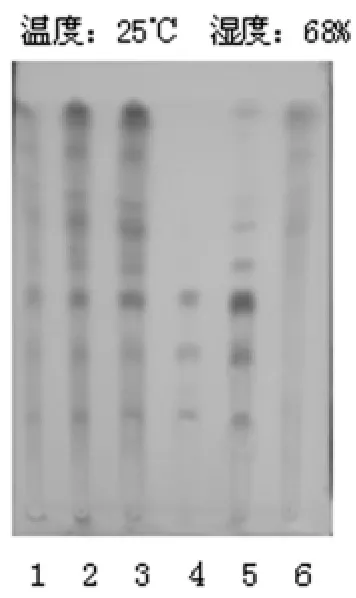
圖2
2.2 含量測定
2.2.1 色譜條件與系統適應性試驗 色譜柱:Agilent ZORBAX C18 柱(250 mm ×4.6 mm,5μm);流動相:甲醇-水(73∶27);檢測波長:270nm;柱溫:30℃;理論板數按丹參酮ⅡA峰計算應不低于2000,進樣量10 μl。該條件丹參酮ⅡA分離度好,丹參陰性對照無干擾[2]。色譜圖見圖3。
2.2.2 對照品溶液的制備 取丹參酮ⅡA對照品約10 mg,精密稱定,置50 ml棕色容量瓶中,加甲醇制成每1 ml含40 μg的溶液,即得[3]。
2.2.3 供試品溶液的制備 取本品適量研細,取約1 g,精密稱定,置具塞棕色瓶中,精密加入甲醇25 ml,密塞,稱定重量,超聲處理(功率205W,頻率33kHz)15 min,放冷,再稱定重量,用甲醇補足減失的重量,搖勻,濾過,取續濾液,置棕色瓶中,即得[4-6]。
2.2.4 陰性對照溶液的制備 取相當于供試品量1 g的缺丹參陰性對照樣品,同法制成丹參陰性對照溶液。
2.2.5 標準曲線的制備 精密吸取對照品溶液1、2、5、10、15、20 μl,依次進樣測定。以質量Y為縱坐標,峰面積X為橫坐標,繪制標準曲線,并進行線性回歸。其回歸方程和相關系數為:Y=227.22X+1.3829,r=0.9999,丹參酮ⅡA在0.04032~0.8064 μg范圍內與峰面積線性關系良好。
2.2.5 精密度試驗 精密吸取同一份對照品溶液10 μl,重復測定6次,丹參酮ⅡA的峰面積RSD為0.55%。
2.2.6 重復性試驗 取同一批樣品(20081129)6份,分別按“2.2.3”項下方法制備,精密吸取10 μl進樣,測得樣品中丹參酮ⅡA平均含量0.52,RSD為0.74%。
2.2.7 穩定性試驗 精密吸取同一份樣品溶液10 μl,分別0、2、4、6、8、10 h 測定,RSD 為 0.59%,說明被測溶液在 10 h內基本穩定。
2.2.8 加樣回收試驗 取已知含量三高調和丸的樣品粉末約0.5 g,精密稱取共6份,置棕色瓶中,分別精密加入丹參酮ⅡA對照品溶液25 ml,按2.2.3項下方法制備,分別測定,計算回收率,結果見表1。
2.2.9 樣品測定 取三高調和丸樣品3批,按2.2.3項下方法制備供試品溶液,按上述色譜條件測定。結果見表2。

圖3 HPLC色譜圖
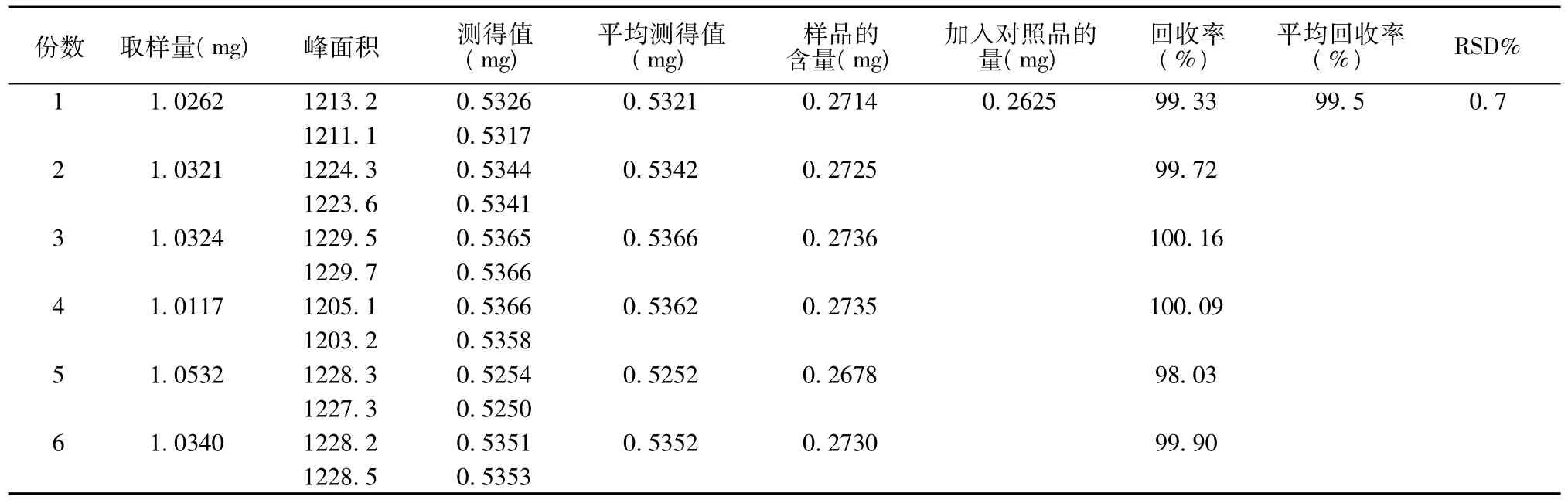
表1 加樣回收率試驗結果

表2 樣品測定結果
4 討論
4.1 丹參和三七的薄層色譜鑒別操作簡單,結果斑點對應性好,陰性對照均無干擾,具有專屬性。在不同溫度10℃、20℃、30℃及不同濕度,不用同廠家硅膠G薄層板實驗結果均一致。
4.2 定量測定參照《中國藥典》2010年版一部復方丹參片的方法,簡便準確。實驗中考察了超聲提取時間5、10、15、30 min的提取效果,結果3個超聲提取時間含量測定結果基本一致,為保證提取完全同時節省實驗時間,最后選擇超聲時間為15 min。
[1] 國家藥典委員會.中國藥典(一部).北京:中國醫藥科技出版社,2010:附錄ⅥB.
[2] 張英鋒,王燕革,馬子川,等.丹參活性化學成分的研究.化學世界,2009,50(10)
[3] 邱麗麗,容蓉,蔣海強,等.高效液相色譜法同時測定丹參提取物中4種成分的含量.分析試驗室,2009,28(21).
[4] 藍天鳳.丹參中丹參酮類成分及其含量測定方法研究.山東中醫藥大學,2011.
[5] 楊東風.丹參藥材HPLC指紋圖譜及其質量評價研究.西北農林科技大學,2007.
[6] 曹冬.中藥丹參及其制劑的質量控制方法研究.河北醫科大學,2006.
