固相萃取/超高效液相色譜-電噴霧串聯(lián)質(zhì)譜法檢測酶制劑中3-硝基丙酸
2013-10-08 00:51:54樂愛山孔祥虹
分析測試學(xué)報(bào) 2013年6期
關(guān)鍵詞:實(shí)驗(yàn)
張 璐,樂愛山,鄭 玲,孔祥虹
(1.陜西出入境檢驗(yàn)檢疫局,陜西 西安 710068;2.廣西出入境檢驗(yàn)檢疫局,廣西 南寧 530022)
酶制劑是果汁加工過程中廣泛使用的加工助劑[1-3],常用的有淀粉酶、果膠酶、蛋白酶、糖化酶、脂肪酶、超濾酶等。果汁生產(chǎn)中使用多種酶制劑以提高加工效率,增加產(chǎn)品穩(wěn)定性,如使用淀粉酶來轉(zhuǎn)化未成熟果實(shí)中的淀粉,使用果膠酶以除去榨汁中的果膠,使用超濾酶增加果汁超濾時(shí)的通過率。但用米曲霉發(fā)酵產(chǎn)生的酶制劑可能含有強(qiáng)毒物質(zhì)3-硝基丙酸(3-Nitropropionic acid,3-NPA)。3-硝基丙酸是一種無色針狀晶體,為高等植物受霉菌污染產(chǎn)生的有毒物質(zhì),可引起牲畜中毒,其中毒癥狀主要表現(xiàn)為中樞神經(jīng)系統(tǒng)的損害[4-7],急性期表現(xiàn)有嘔吐、眩暈、陣發(fā)性抽搐、昏迷等,嚴(yán)重可導(dǎo)致死亡。聯(lián)合圍糧農(nóng)組織和世界衛(wèi)生組織關(guān)于食品添加劑的聯(lián)合專家委員會(Joint FAO/WHO Expert Committee on Food Additives)在1987即明確規(guī)定用米曲霉發(fā)酵生產(chǎn)的淀粉酶、蛋白酶、葡萄糖化酶、脂肪酶等時(shí)均要檢測3-NPA。
目前對于3-NPA的檢測主要集中在甘蔗及甘蔗制品[8-9],且檢測方法多為高效液相色譜法[10]、氣相色譜法[11]、薄層色譜法[8]等。但這些方法均存在樣品前處理繁瑣、需使用大量毒性大的有機(jī)試劑、干擾較大或需要進(jìn)行衍生等問題。本文通過對提取、凈化、測定及確證等條件的研究及優(yōu)化,建立了檢測酶制劑中3-NPA的固相萃取/超高效液相色譜電噴霧串聯(lián)質(zhì)譜方法,可實(shí)現(xiàn)對3-NPA的定性及定量分析。
1 實(shí)驗(yàn)部分
1.1 儀器、試劑與材料
Waters ACQULTY UPLC-Quattro Premier XE液相色譜-質(zhì)譜聯(lián)用儀(美國Waters公司);冷凍離心機(jī)(美國Beckman公司);漩渦混合儀(德國IKA公司);LABOROTA4001旋轉(zhuǎn)濃縮儀(德國Heidolph公司);Milli-Q超純水儀(美國Millipore公司)。
3-NPA(97.5%,Dr.Ehrenstorfer公司);乙腈(色譜純,F(xiàn)isher公司);氯化鈉、無水硫酸鈉均為分析純,無水硫酸鈉使用前于600℃馬弗爐中烘烤5 h后密封保存。PSA固相萃取柱(3 mL,500 mg,Supelco公司)。
3-NPA標(biāo)準(zhǔn)儲備液的配制:稱取標(biāo)準(zhǔn)品10.0 mg于10 mL棕色容量瓶,用甲醇溶解并定容至10 mL,配成1.0 g/L的標(biāo)準(zhǔn)儲備液,于-20℃下保存。3-NPA標(biāo)準(zhǔn)工作溶液的配制:取上述儲備液0.025 mL于棕色容量瓶中,用甲醇定容至25 mL,配制成1.0 mg/L的標(biāo)準(zhǔn)工作溶液。
1.2 樣品前處理
1.2.1 液體酶制劑中3-NPA的提取 稱取5.00 g酶制劑樣品于50 mL離心管中,加入20 mL乙腈振蕩20 min后,再加入5 g氯化鈉混合均勻后振蕩10 min,以4 500 r/min離心5 min。取上清液10 mL,待凈化。
1.2.2 固體酶制劑中3-NPA的提取 稱取5.00 g酶制劑樣品于50 mL離心管中,加入20 mL乙腈振蕩20 min后,以4 500 r/min離心5 min。取上清液10 mL,待凈化。
1.2.3 凈 化 PSA固相萃取柱上加1.0 g無水硫酸鈉后用6 mL乙腈活化,將試樣溶液過柱,用10 mL甲醇洗脫,收集洗脫液。經(jīng)氮?dú)獯抵两珊螅?.5% 甲酸水溶液定容至1.0 mL,過0.22 μm濾膜后,待測。
1.3 儀器條件
色譜條件:色譜柱Waters HSS T3柱(1.8 μm,2.1 mm×100 mm)。流動相為乙腈(A)和水(B),梯度洗脫程序:0~0.5 min,3% ~10%A;0.5~2.0 min,10% ~80%A;2.0~2.8 min,80%A;2.8~3.5 min,80% ~3%A;3.5~4.5 min,3%A。流速0.3 mL/min,進(jìn)樣體積5.0 μL,柱溫40℃。
MS/MS條件:離子源為電噴霧電離源,負(fù)離子模式(ESI-),毛細(xì)管電壓為2.91 kV;離子源溫度115℃;脫溶劑氣溫度:450℃;脫溶劑氣流量500 L/h;碰撞氣為氬氣,流量為0.2 mL/min;錐孔電壓13 V;碰撞能量10 eV;檢測模式為多反應(yīng)監(jiān)測(MRM);母離子為m/z117.9;特征離子為m/z46.3。
2 結(jié)果與討論
2.1 前處理?xiàng)l件的優(yōu)化
考察了甲醇、乙腈、乙酸乙酯等不同有機(jī)溶劑的提取效果。結(jié)果表明,甲醇的提取率為90%但干擾較大,乙酸乙酯的提取率僅為70%,而乙腈的提取率達(dá)到95%且干擾較少。同時(shí)在提取過程中加入氯化鈉將水相和有機(jī)相分離。適量的氯化鈉在離心后加入有助于減少有機(jī)溶劑中的漂浮物,使樣品更清澈,有利于固相萃取的進(jìn)行。
2.2 固相萃取柱的選擇
據(jù)文獻(xiàn)報(bào)道[12],可采用乙酸乙酯和三氯甲烷液液萃取的方式提取凈化甘蔗中的3-NPA。但該方法的溶劑使用量大,且多次萃取容易造成待測物損失。本研究中采用固相萃取技術(shù)完成目標(biāo)化合物的富集及凈化。實(shí)驗(yàn)考察了不同萃取柱(Oasis WAX、PSA及氨基柱)的凈化效果,結(jié)果顯示,對酸性物質(zhì)有較好選擇性的Oasis MAX柱[13]雖能將目標(biāo)物完全保留在小柱上,但無合適的洗脫液將目標(biāo)物洗脫下來。
實(shí)驗(yàn)中比較了特征相似的氨基柱和PSA柱的凈化效果。使用加標(biāo)的乙腈溶液上固相萃取柱后,收集上柱溶液進(jìn)行分析,3-NPA完全保留在氨基柱和PSA柱上。研究表明當(dāng)強(qiáng)極性化合物在氨基柱上吸附力太強(qiáng)時(shí)可考慮用PSA柱代替[14]。比較了3-NPA的乙腈溶液在氨基柱和PSA柱上的解吸能力,在實(shí)驗(yàn)中用不同的洗脫液分別洗脫吸附在兩種固相萃取柱上的3-NPA(見圖1)。實(shí)驗(yàn)結(jié)果表明,過PSA固相萃取柱的回收率高于氨基柱,可能是由于目標(biāo)化合物3-NPA在氨基柱上吸附過強(qiáng)。因此實(shí)驗(yàn)選擇PSA柱為固相萃取柱進(jìn)行進(jìn)化。
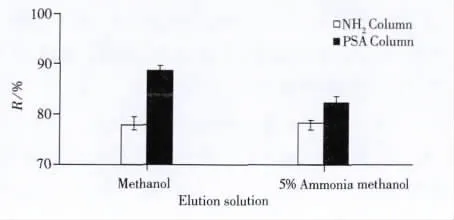
圖1 3-NPA在不同固相萃取柱上的解離圖Fig.1 Figure of dissociation of 3-NPA in different solid phase extraction columns
2.3 PSA固相萃取條件的優(yōu)化
為了提高凈化效果和降低基質(zhì)效應(yīng),一般會對固相萃取柱進(jìn)行淋洗。實(shí)驗(yàn)考察了不同體積比(10∶90、12∶88、20∶80)的異丙醇-乙腈作為淋洗液時(shí)3-NPA的損失情況。結(jié)果表明,用上述淋洗液后3-NPA均有損失。因此實(shí)驗(yàn)不采用淋洗步驟,而通過基質(zhì)標(biāo)準(zhǔn)曲線降低基質(zhì)效應(yīng)的影響。
吸附于PSA固相萃取柱上的目標(biāo)化合物,需要強(qiáng)極性溶劑進(jìn)行洗脫。實(shí)驗(yàn)考察了乙腈、甲醇以及不同體積比的乙腈-甲醇(50∶50、30∶70、10∶90)溶液的洗脫能力,實(shí)驗(yàn)結(jié)果表明,甲醇比例越高,吸附在PSA柱上的3-NPA越容易洗脫下來。最終選擇100%甲醇作為洗脫溶液。進(jìn)一步考察了甲醇用量(3、5、8、10、15、20 mL)的影響,結(jié)果顯示當(dāng)洗脫溶液體積達(dá)到10 mL時(shí),3-NPA已被全部洗脫下來,最終確定10 mL甲醇為洗脫溶液。
2.4 色譜柱的選擇
文獻(xiàn)報(bào)道使用反相色譜柱分離3-NPA時(shí),由于3-NPA具有很強(qiáng)的酸性和親水性,在反相柱上保留較弱,因此需要使用磷酸二氫鉀溶液作為流動相[15]。由于液質(zhì)聯(lián)用不建議用磷酸鹽等不揮發(fā)性物質(zhì)為流動相,因此實(shí)驗(yàn)重點(diǎn)考察了有利于極性物質(zhì)保留的反相色譜柱Waters ACQUITY HSS T3、Waters ACQUITY C18、Aglient Eclipse Plus C18、Waters UPLC BEH Amide及親水性色譜柱Waters ACQUITY BEH HILIC對3-NPA的分離效果(見圖2)。結(jié)果表明,3-NPA在Waters ACQUITY C18及Aglient Eclipse Plus C18的保留時(shí)間均在0.7 min,且峰形拖尾,在Waters ACQUITY BEH HILIC及Waters UPLC BEH Amide上的峰形盡管有所改善但仍有拖尾。而3-NPA在Waters ACQUITY HSS T3上的保留時(shí)間在1.5 min,且峰形良好。因此選擇Waters ACQUITY HSS T3作為3-NPA的分析柱。
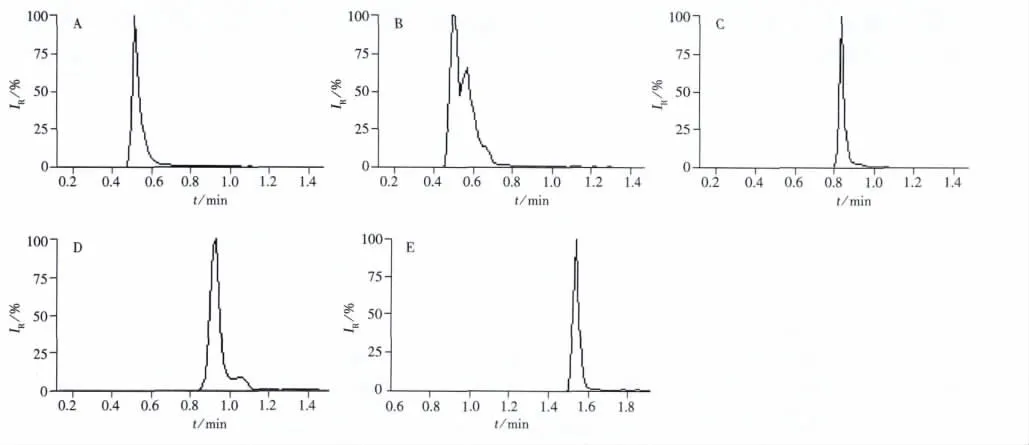
圖2 3-NPA在不同色譜柱上的保留色譜圖Fig.2 Chromatograms of retention of 3-NPA on different columns A.Waters ACQUITY C18(1.8 μm,2.1 mm ×100 mm);B.Aglient Eclipse Plus C18(1.8 μm,2.1 mm ×100 mm);C.ACQUITY BEH HILIC(1.8 μm,2.1 mm ×100 mm);D.Waters UPLC BEH Amide(1.7 μm,2.1 mm ×100 mm);E.Waters ACQUITY HSS T3(1.8 μm,2.1 mm ×100 mm)
2.5 流動相條件的優(yōu)化
由于3-NPA是一種小分子有機(jī)酸,具有較強(qiáng)的酸性和親水性。因此主要考察了甲醇-水、乙腈-水、乙腈-5 mmol/L乙酸銨溶液體系的分離效果。結(jié)果表明,乙腈-水為流動相時(shí)的響應(yīng)最高,且保留時(shí)間在1.5 min以后。流動相中加鹽后靈敏度降低,可能是鹽的存在抑制了目標(biāo)化合物的電離。因此確定乙腈-水為最佳流動相。
為了增強(qiáng)目標(biāo)化合物在色譜柱上的保留。實(shí)驗(yàn)比較了純水、0.1%氨水溶液、0.05%甲酸水溶液、0.5%甲酸水溶液、5 mmol/L甲酸銨溶液作為定容液時(shí)對目標(biāo)化合物響應(yīng)的影響。實(shí)驗(yàn)發(fā)現(xiàn),用0.5%甲酸水溶液作為定容溶液時(shí)不僅保留時(shí)間明顯增強(qiáng),且響應(yīng)也明顯增強(qiáng)。最終確定0.5%甲酸水溶液作為定容溶液。
2.6 基質(zhì)干擾的影響
由于酶制劑大多由真菌菌種發(fā)酵而成,含有大量蛋白,因此對3-NPA的提取及測定有干擾,且無法得到3-NPA的同位素內(nèi)標(biāo)。因此選擇基質(zhì)加標(biāo)工作曲線進(jìn)行外標(biāo)法定量,對基質(zhì)效應(yīng)及凈化過程中的損失進(jìn)行校正。以果膠酶為例,不采用果膠酶基質(zhì)標(biāo)準(zhǔn)時(shí),加標(biāo)5.0 μg/kg的回收率為32.1%~42.1%,而采用基質(zhì)標(biāo)準(zhǔn)后,回收率為63.2%~79.6%。回收率提高了1倍。實(shí)驗(yàn)還比較了不同酶制劑為基質(zhì)標(biāo)準(zhǔn)對3-NPA檢測的干擾校正情況(見圖3),不同酶制劑對3-NPA的干擾影響不同,果膠酶對3-NPA的抑制干擾最大,果膠超濾酶的干擾較小,因此實(shí)驗(yàn)需要對不同品種的酶制劑進(jìn)行基質(zhì)標(biāo)準(zhǔn)校正,進(jìn)而進(jìn)行定量分析。
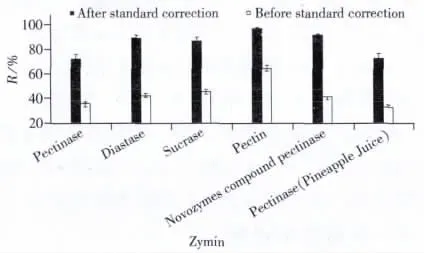
圖3 不同酶制劑的基質(zhì)影響效應(yīng)Fig.3 The matrix effects of different zymins
2.7 方法驗(yàn)證參數(shù)
對不同種類的酶制劑繪制基質(zhì)標(biāo)準(zhǔn)曲線。分別稱取6份5.0 g空白酶制劑基質(zhì)于50 mL離心管中,分別加入1.0 μg/L標(biāo)準(zhǔn)工作溶液25、50、100、150、250 μL,進(jìn)行前處理后在優(yōu)化條件下測定。以3-NPA的峰面積(Y)對相應(yīng)的質(zhì)量濃度(X,μg/L)繪制基質(zhì)標(biāo)準(zhǔn)工作曲線。得出回歸方程、相關(guān)系數(shù)、線性范圍及定量下限(S/N≥10)(見表1)。結(jié)果顯示,3-硝基丙酸在6種酶制劑基質(zhì)中的線性范圍均為12.5 ~125 μg·L-1,相關(guān)系數(shù)大于 0.993,定量下限為 5.0 μg/kg。
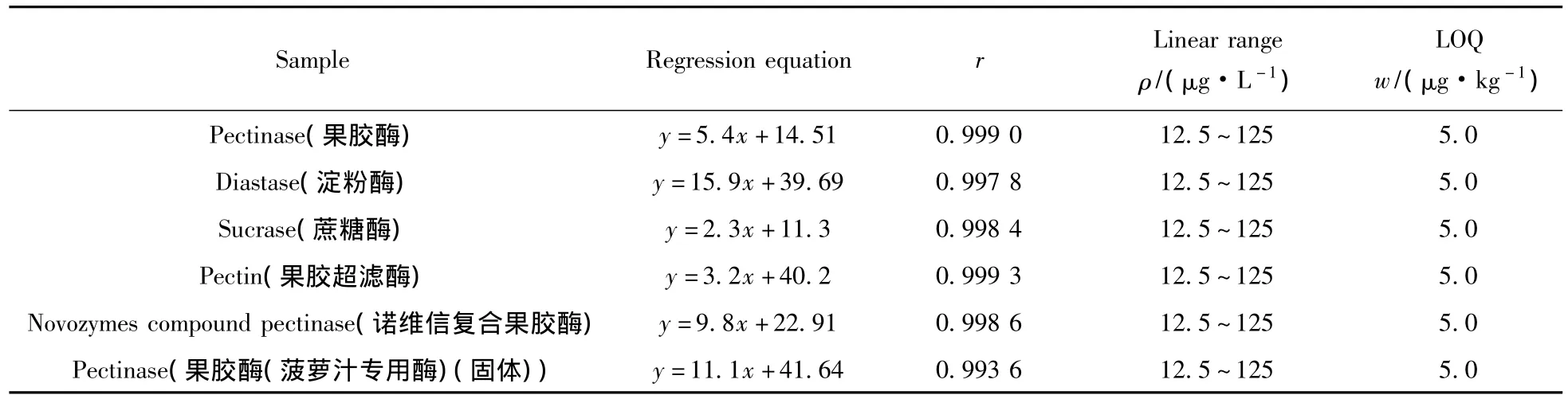
表1 不同酶制劑的回歸方程、相關(guān)系數(shù)(r)、線性范圍及定量下限Table 1 Regression equation,correlation coefficient(r),linearity range and detection limit in different zymin
分別稱取不含3-NPA的空白果膠酶、果膠超濾酶、諾維信復(fù)合果膠酶、蛋白酶、蔗糖酶樣品進(jìn)行方法回收率和精密度試驗(yàn)。加入的3-NPA標(biāo)準(zhǔn)品濃度分別為5.0、10.0、20.0 μg/kg,每種濃度做5次平行,按優(yōu)化方法進(jìn)行實(shí)驗(yàn)。每種樣品的平均回收率及相對標(biāo)準(zhǔn)偏差(RSD)見表2。在3個(gè)加標(biāo)水平下,3-硝基丙酸的回收率為69.1%~114.4%,RSD為6.9%~8.9%。
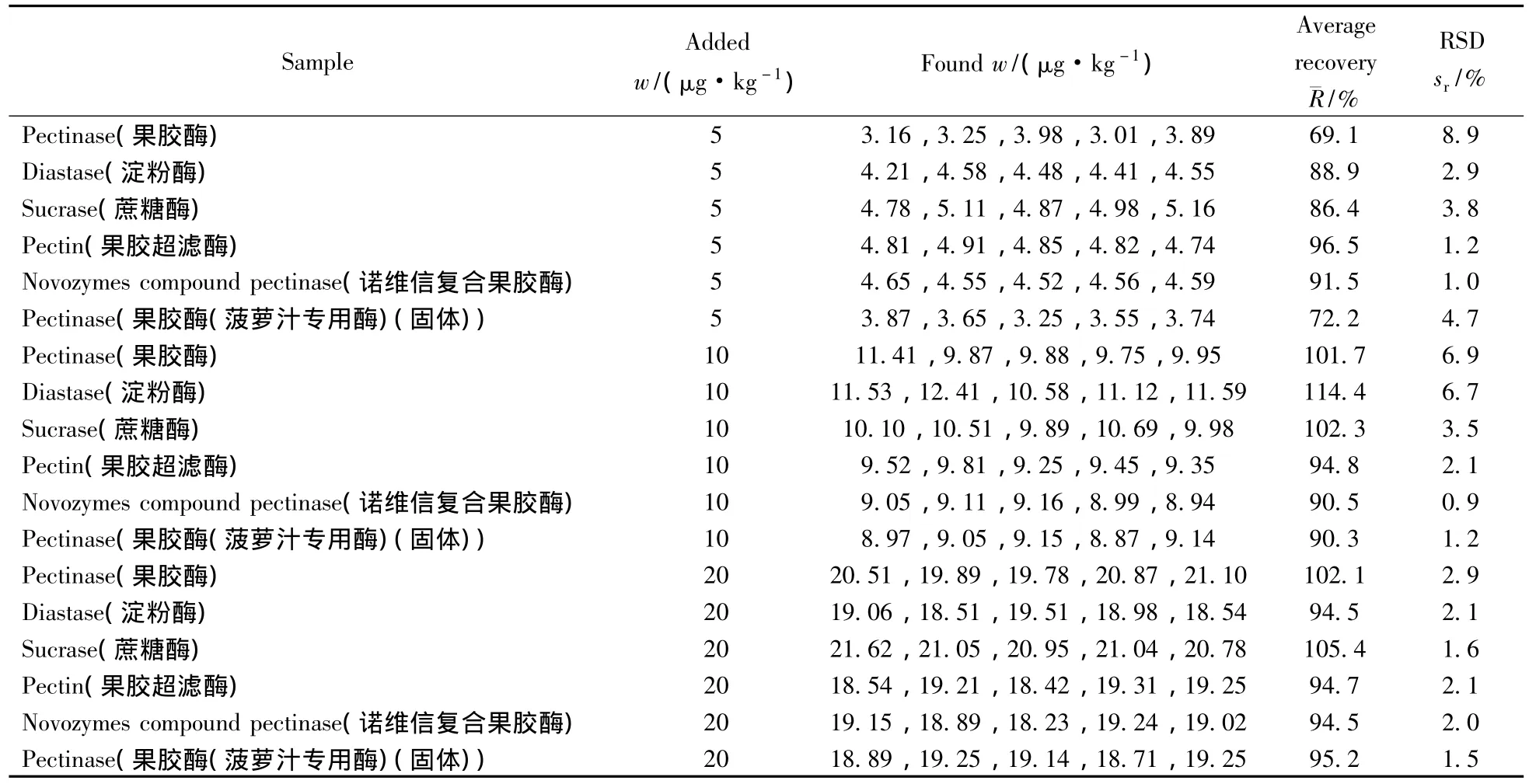
表2 3-NPA在不同種類酶制劑的加標(biāo)回收率及相對標(biāo)準(zhǔn)偏差Table 2 Recovery and precision of 3-NPA in different enzymic preparations
2.8 方法應(yīng)用
為了考察本方法的有效性與實(shí)用性,采購7種常用的酶制劑樣品進(jìn)行3-NPA的檢測。結(jié)果表明,所有樣品中均未檢出3-NPA殘留。圖4為酶制劑實(shí)際樣品的色譜圖。
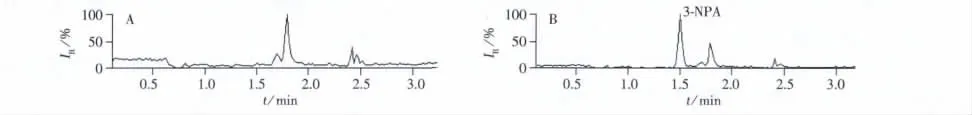
圖4 果膠酶樣品(A)及樣品加標(biāo)(B)的色譜圖Fig.4 Chromatograms of pectinase sample(A)and sample spiked with 3-NPA(B)
3 結(jié)論
本文建立了酶制劑中3-NPA的超高效液相色譜-電噴霧串聯(lián)質(zhì)譜檢測方法。樣品經(jīng)乙腈提取,固相萃取柱凈化,質(zhì)譜確認(rèn)后,可實(shí)現(xiàn)對3-NPA的定性及定量分析。該方法前處理過程簡便、靈敏度高、選擇性好,可滿足日常檢測的需要,為酶制劑的質(zhì)量安全提供了技術(shù)保障。
[1] Leza H A R,Jasso R M R,Esquivel J C C,Herrera R R,Aguilar C N.Biohem.Eng.J,2012,65:90-95.
[2] Carrín M E,Ceci L N,Lozano J E.Food Chem.,2004,84:173 -178.
[3] Tribst A A L,Cristianini M.Innovative Food Science and Emerging Technologies,2012,13:107 -111.
[4] Li F,Xu Q Y.Lab.Animal Sci.(李峰,徐群淵.實(shí)驗(yàn)動物科學(xué)),2009,26(4):12-15.
[5] Liu H G,Ma Y,Yang A C,Meng D W,Zhang Y,Zhang J G.Chinese Journal of Minimally Invasive Neurosurgery(劉煥光,馬羽,楊岸超,孟大偉,張穎,張建國.中國微侵襲神經(jīng)外科雜志),2012,17(7):319-321.
[6] Olsen C,Rustad A,F(xiàn)onnum F,Paulsen R E,Hassel B.Brain Res.,1999,850:144 -149.
[7] Luchowski P,Luchowska E,Turskia W A,Urbanskaa E M.Neurosci.Lett.,2002,330:49 -52.
[8] Liu Y,Wang Y H,Liu X J,Li X F,Liu Z H,Hu W J.J.Hyg.Res.(劉勇,王玉華,劉興玠,李秀芳,劉增輝,胡文娟.衛(wèi)生研究),1989,18(5):38-40.
[9] Shao G J,Han J K,Wu D Q.Chin.J.Health Lab.Technol.(邵國健,韓建康,吳丹青.中國衛(wèi)生檢驗(yàn)雜志),2012,22(4):711-712.
[10] Jiang T,Zhang Q L,Luo X Y.J.Hyg.Res.(江濤,張慶林,羅雪云.衛(wèi)生研究),1999,28(5):300-302.
[11] Wang J,Lei Z Y,F(xiàn)eng X Q.Pratacultural Science(汪儆,雷祖玉,馮學(xué)勤.草業(yè)科學(xué)),1992,9(2):34-37.
[12] WS/T 10 -1996.Diagnotic Criteria and Principles of Management for Food Poisoning of Mildew Sugareane.Standards of Ministry of Health of the Peoples Republic of China(變質(zhì)甘蔗食物中毒診斷標(biāo)準(zhǔn)及處理原則.中華人民共和國衛(wèi)生部標(biāo)準(zhǔn)).
[13] Li B,Wu G H,Liu W,Zhao X D,Zhao H Y,Xue Y,Zhao R.Chin.J.Food Hyg.(李兵,吳國華,劉偉,趙旭東,趙海燕,薜穎,趙榕.中國食品衛(wèi)生雜志),2012,24(2):127-132.
[14] Chen X H,Wang Q J.Technology and Application of Solid-phase Extraction.Beijing:Science Press(陳小華,汪群杰.固相萃取技術(shù)與應(yīng)用.北京:科學(xué)出版社),2010:56.
[15] Gong X M,Ren Y P,Dong J,Sun J,Li J,Jin C,Yu J L.J.Instrum.Anal.(宮小明,任一平,董靜,孫軍,李健,金超,于金玲.分析測試學(xué)報(bào)),2011,30(1):6-12.
猜你喜歡
作文·小學(xué)低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學(xué)生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學(xué)低年級(2024年2期)2024-04-29 00:00:00
作文·小學(xué)低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(bào)(2022年4期)2022-08-09 08:52:06
中學(xué)生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學(xué))(2019年6期)2019-10-10 01:01:50
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
