反應型非離子聚氨酯表面活性劑的合成及性能
2013-10-27 02:28:12劉娟娟
天津工業大學學報 2013年1期
劉娟娟,呂 彤
(天津工業大學環境與化學工程學院,天津 300387)
反應型表面活性劑帶有活性基團,它除了具有優良的表面活性之外,還可以通過化學鍵的作用鍵合在粒子表面,參與聚合反應,從而解決了傳統表面活性劑對產品和環境帶來的問題[1-3].聚氨酯表面活性劑不僅能夠顯著降低表面張力,而且由于聚氨酯分子結構易裁剪,因而具有優良的分子結構可調控性和生物相容性,故而成為目前一類備受歡迎的新型高分子表面活性劑[4-6].目前對于聚氨酯表面活性劑的研究主要集中在陰離子性表面活性劑上,如Zhang等[7]和朱再盛等[8]的研究,但這種離子型的表面活性劑由于離子基團的存在,易受酸、堿、鹽等電解質的影響,因此近幾年有研究小組致力于合成非離子聚氨酯表面活性劑.陳大俊等[9]用聚乙二醇、4,4-二苯基甲烷二異氰酸酯和甲基丙烯酸羥乙酯為原料制備了兩端都含有雙鍵的反應型表面活性劑,具有優良的聚合性能,但其分子中含有過多的疏水鏈段,濁點較低,在一定程度上限制了其應用范圍;胡應模等[10]以甲苯二異氰酸酯、馬來酸酐和聚乙二醇和三乙胺為主要原料合成了兼有非離子和陰離子性能的可聚合聚氨酯型表面活性劑,具有較好的乳化能力,但由于立體位阻的影響難以均聚或共聚形成聚合物,且由于其結構中親水基團太長,臨界膠束濃度數量級只有10-2;張彪等[11]以異佛爾酮二異氰酸酯、聚乙二醇單甲醚、聚氧丙烯和丙烯酸羥乙酯為主要原料,通過逐步聚合,合成了含有PEG/PPO鏈段反應型聚氨酯表面活性劑,這種表面活性劑相容性好,但反應過程非常復雜,產品純度難于控制,且分子結構中含有大量的疏水基團,HLB值較低,水溶性必定不高.因此,合理設計表面活性劑的親水鏈段/疏水鏈段的比例[12],是探索合成反應活性高、應用范圍廣的反應型表面活性劑的關鍵.本文通過設計首先以聚乙二醇和甲基丙烯酸為原料,酯化得到甲基丙烯酸聚乙二醇單酯(引入雙鍵),然后與異佛爾酮二異氰酸酯反應,最后用烷基醇(碳數為4、8和12)封端,合成出一系列的反應型非離子聚氨酯表面活性劑,探索其合成的工藝條件,并對目標產物的結構和物化性能進行了研究.這種表面活性劑的結構容易調整,可根據需要的親水親油平衡值選擇合適碳原子數的聚乙二醇和烷基醇,具有較高的實用的價值.
1 實驗部分
1.1 試劑與儀器
試劑:聚乙二醇(PEG1000),分析純,上海浦東高甫化工廠生產;甲苯、對甲苯磺酸,分析純,天津市化學試劑三廠生產;二月桂酸二正丁基錫(DBTDL)、過硫酸鉀(KPS),化學純,天津市華東試劑廠生產;二正丁胺,分析純,天津市福晨化學試劑廠生產;異佛爾酮二異氰酸酯(IPDI),分析純,德國Bayer公司生產;正丁醇、正辛醇、十二醇,分析純,均為天津市博迪化工有限公司生產;甲基丙烯酸甲酯(MMA)、十二烷基磺酸鈉(SDS)、丙烯酸丁酯(BA),分析純,天津市光復精細化工研究所生產;三氯甲烷,分析純,天津市風船化學試劑科技有限公司生產.
儀器:TENSOR37型傅里葉紅外吸收光譜儀,德國Bruker公司生產;300 MHz傅里葉變換超導核磁共振譜儀,德國Bruker公司生產;QBZY型全自動表面張力儀,上海方瑞有限公司生產.
1.2 合成方法
在裝有攪拌器、冷凝管、分水器和溫度計的三口瓶中,加入PEG和阻聚劑對苯二酚,加熱并攪拌,待反應溫度上升到60~70℃時加入催化劑對甲苯磺酸和脫水劑甲苯,繼續升高溫度至85~90℃.將MAA慢慢滴加到反應液中,迅速升溫至110~120℃,反應7 h,用氯仿溶解,用飽和NaHCO3、飽和食鹽水洗滌,除去催化劑、阻聚劑、未反應的甲基丙烯酸和聚乙二醇.分液,將有機相減壓蒸餾除去氯仿、甲苯,得到純化的甲基丙烯酸聚乙二醇單酯(PMA).
將一定計量的IPDI及適量的DBTDL加入帶有攪拌器和溫度計的三口燒瓶中,添加丙酮后進行攪拌使其混合均勻,并通氮氣保護,10 min以后開始慢慢滴加提純的PMA,溫度控制在40~60℃反應2 h.再加入正丁醇(正辛醇、十二醇),反應2~3 h.反應完成后用石油醚沉淀,重復3次除去未反應原料,真空干燥,得最終反應型非離子聚氨酯表面活性劑產品,記PU1、PU2和PU3.
1.3 結構表征及性能測試
(1)紅外光譜:采用涂膜法進行IR光譜測試,將制成的表面活性劑涂膜在溴化鉀鹽片上,常溫下掃描.
(2)核磁共振:用氘代氯仿溶解,進行1H譜測試.
(3)表面張力測定方法:配制一系列不同濃度的水溶液,采用全自動表面張力儀于25℃測定.
(4)乳化能力的測試:將5 mL待測的單體(MMA、BA)、10 mL質量分數為1.0%的表面活性劑水溶液置于具塞量筒中,然后用力上下震蕩10次,停10 s,再震蕩10次,重復3次后,靜置2 h,以析出水的體積(mL)衡量其乳化穩定性.
(5)濁點的測試:配制15 mL質量分數為1%的非離子表面活性劑水溶液,水浴中慢慢加熱并不斷攪拌,觀察液體開始變渾濁時的溫度,取出試管,冷卻,再觀察混濁液變澄清時的溫度,重復3次,取其平均值作為該表面活性劑的濁點.
2 結果與討論
2.1 反應條件對PMA合成的影響
本文首先通過單因素實驗初步考察了投料比nPEG∶nMAA(A)、催化劑用量(以 PEG 計的質量分數)(B)、阻聚劑(以MAA計的質量分數)(C)及反應溫度(D)這4個因素對合成的影響.在此基礎上優化實驗條件,選定以酯化率為指標,進行了4因素3水平正交實驗.正交實驗結果見表1.
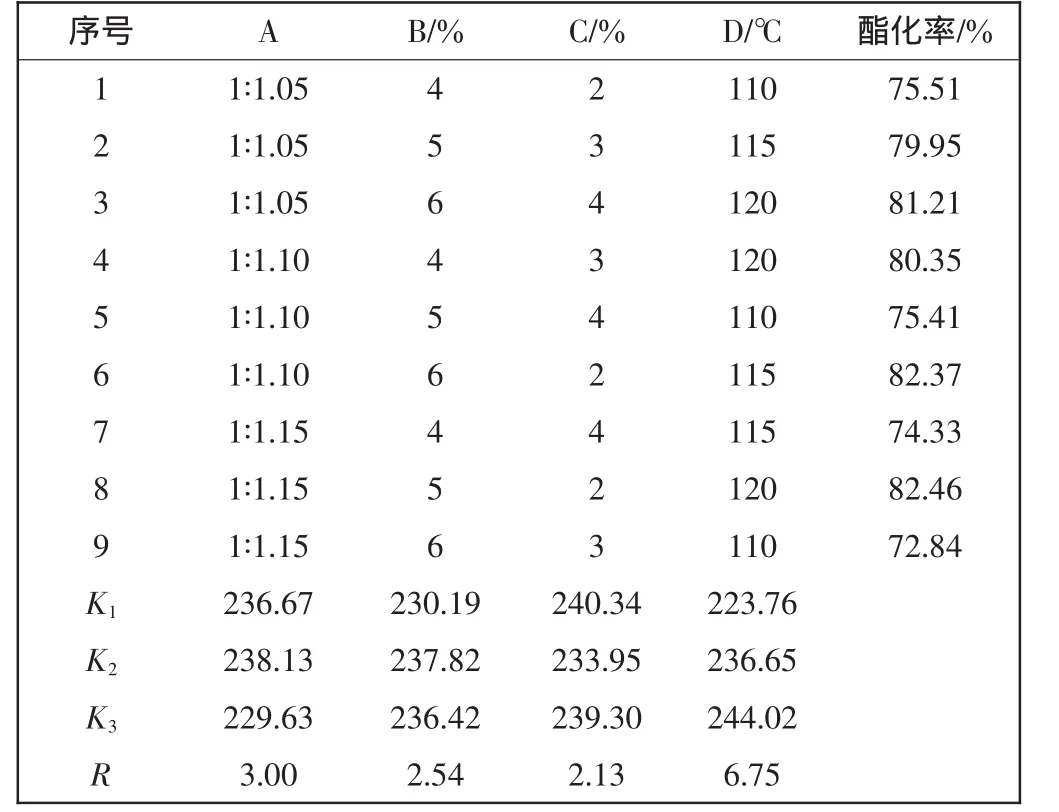
表1 正交試驗結果Tab.1 Results of the orthogonal test
由表1中極差分析結果可知,水平效應值R的大小決定了各因素影響的大小順序為:D>A>B>C.最佳酯化條件為:nPEG∶nMMA=1∶1.10,對甲苯磺酸質量分數為5%,對苯二酚質量分數為2%,反應溫度為120℃.在此條件下,重復試驗進行驗證,酯化率可達78.5%以上.
2.2 反應條件對表面活性劑合成的影響
IPDI中2個NCO基團分別位于脂肪族的伯位和脂環族的仲位,2個NCO基團的活性差在0.2∶1~12∶1的范圍內[13].伯異氰酸酯基與亞甲基相連,仲脂環異氰酸酯基與環相連,因伯異氰酸酯基被β位置上的甲基取代物環己烷環和它的相鄰的甲基有效地保護起來,故仲脂環異氰酸酯基的活性大于伯異氰酸酯基的活性.根據文獻[14]通過改變催化劑或反應溫度,可使2個NCO基團的反應活性差異程度變大,使活性基團在第一步優先與仲位的NCO反應.
2.2.1 反應溫度的影響
反應溫度對表面活性劑合成的影響如圖1和圖2所示.
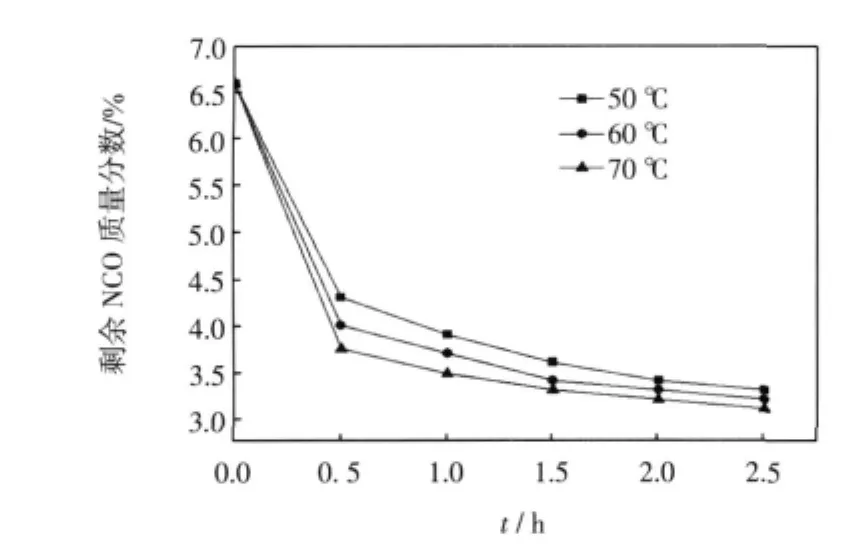
圖1 溫度對第1步反應的影響Fig.1 Effect of temperature on first step
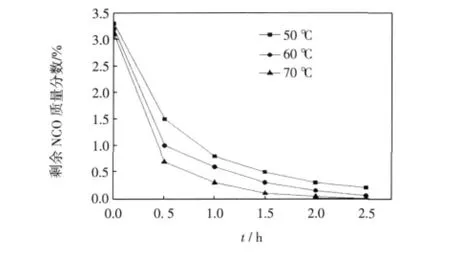
圖2 溫度對第2步反應的影響Fig.2 Effect of temperature on second step
對于第一步IPDI與PMA的反應,以DBTDL為催化劑,考察在不同的反應溫度NCO的轉化率隨反應時間的變化情況.從圖1可看出,反應溫度越高,NCO含量下降越快,說明溫度升高有利于反應的進行.但是,分子運動速率會隨著溫度的升高而加快,會使IPDI中2個NCO基團的反應活性差變小,從而導致2個NCO基團的反應選擇性降低.且DBTDL在40~60℃具有很好的催化效果,故選擇60℃左右進行反應較為適宜.由圖2可見,對于第二步與烷基醇的反應,溫度越高,反應越易達到終點.由于脂肪族異氰酸酯基團反應活性較脂環上的異氰酸酯基團低,且已不用考慮溫度對NCO選擇性的影響,可適當提高溫度加快反應,但考慮到溫度過高時會破壞C=C雙鍵,因此溫度控制在65~70℃左右,可保證反應順利且較快地進行.
2.2.2 反應時間的影響
從圖1和圖2也可看出,第1步反應2 h,體系中NCO含量已基本達到理論值,繼續延長反應時間后可能會發生副反應,所以第1步最佳反應時間為2 h.而與烷基醇反應2~2.5 h,NCO含量已經基本達到0,所以第2步最佳的反應時間為2~2.5 h.
2.3 目標表面活性劑的結構表征
2.3.1 紅外光譜(IR)
3種產物的紅外光譜圖基本相同,圖3為PU2表面活性劑樣品的紅外光譜圖.

圖3 PU2的紅外光譜圖Fig.3 FTIR spectroscopy of PU2 surfactant
由圖3可以看出,在3331 cm-1處為氨基甲酸酯結構中N-H的伸縮振動吸收峰,1532 cm-1處為N-H變形振動吸收峰,2870 cm-1處為飽和C-H伸縮振動吸收峰,1718 cm-1處為C=O伸縮振動吸收峰,1638 cm-1處為C=C的吸收峰,1111 cm-1處為聚乙二醇組分C-O-C的特征峰.此外2275 cm-1處的-NCO吸收特征峰消失,說明反應已完成,初步可以斷定實驗得到了預期產物.
2.3.2 氫核磁(1HNMR)波譜分析
圖4所示為PU2表面活性劑樣品的氫核磁譜圖.
1HNMR譜化學位移 δ歸屬為:6.13~5.58[aH,CH2=C(CH3)-],4.21~4.31(bH,-COOCH2-),3.65(cH,-CH2CH2O-)),1.95[dH,CH2=C(CH3)-],1.27~1.55(eh,-CH2CH2-),0.88~1.06(fh,-CH3).以上1H-NMR 數據進一步證實產物為含有活性雙鍵的非離子聚氨酯.
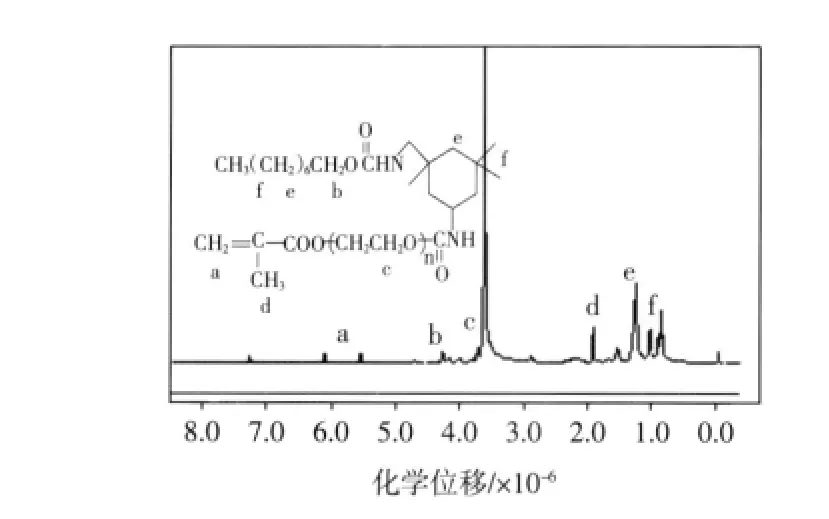
圖4 PU2的1H-NMR譜圖Fig 4 1H-NMR spectroscopy of PU2 surfactant
2.4 表面活性劑的表面張力
圖5為3種產物(PU1、PU2和PU3)的表面張力-濃度的變化曲線.

圖5 PU的表面張力-濃度關系曲線Fig.5 Surface tension versus concentration of PU surfactant
由圖5可以看出,隨著產物質量濃度的增加,溶液表面張力的變化趨勢都是先急速下降,待濃度達到一定值后就基本趨于穩定[15].由此可得PU1、PU2和PU3的臨界膠束質量濃度CMC分別為0.15 g/L(0.109 mmol/L)、0.12g/L (0.085 mmol/L) 和 0.08 g/L(0.054 mmol/L),與之對應的表面張力分別為39.0 mN/m、37.0 mN/m和35.5 mN/m.同時,由3條曲線的對比可以看出,隨著疏水性基團的增加,表面活性劑降低表面張力的能力增強.
2.5 表面活性劑的乳化能力及親水親油平衡值
乳化劑的乳化作用好壞,可以從單體乳液靜置時析出的水量進行判斷,析出的水量越少,乳化能力越強.表2所示為產物對單體的乳化穩定性.
由表2可以看出,制備的表面活性劑對丙烯酸酯類單體有良好的乳化作用,其乳化性能優于傳統的乳化劑SDS,且隨著分子中疏水鏈段的增加,其乳化能力具有明顯增強的趨勢.根據Griffin法計算3種產物的HLB值分別為14.66、14.08和13.54,均在8以上,親水性較好,可以作為O/W型乳化劑.
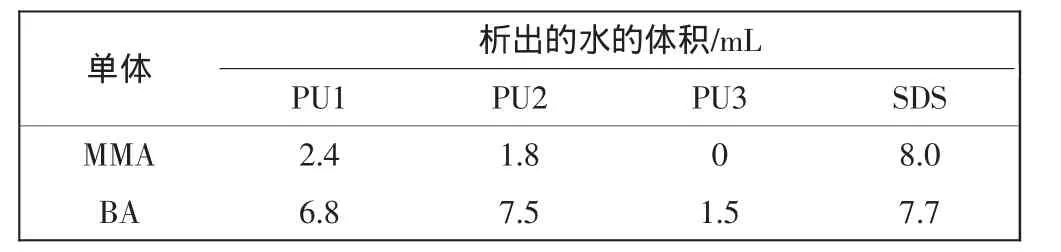
表2 產物對單體的乳化穩定性Tab.2 Stabilities of surfactants to emulsions of monomers
2.6 濁點
濁點是非離子表面活性劑的一特征屬性,當升溫時,由于結合的氫鍵被破壞,使其親水性減弱,因而由原來的透明溶液變成白色混濁的乳濁液.3種產物濁點如表3所示.

表3 產物的濁點Tab.3 Cloud points of surfactants
由表3可以看出,3種產物的濁點隨疏水鏈段含量的增加而降低,這是因為在親水鏈段相同的情況下,親油鏈越長,分子整體與水相的結合力就越弱,越易產生相分離.
2.7 表面活性劑的聚合性能
在裝有攪拌器、溫度計、冷凝管的四口燒瓶中加入表面活性劑(PU3)0.8 g、去離子水80 mL,緩慢滴加混合單體MMA/BA 20 g,在N2的保護下攪拌乳化20 min后,加入引發劑KSP 0.12 g,升溫至75℃反應3 h,于83℃下保溫1 h,得泛藍光的白色乳液.取少量乳液烘烤成膜,得到比較光潔的透明薄膜,說明表面活性劑與單體共聚比較成功.因為丙烯酸酯和聚氨酯共混物相容性較差,在不發生共聚的情況下一般不透明.
3 結論
(1)首先以聚乙二醇、甲基丙烯酸成功合成了甲基丙烯酸聚乙二醇單酯(PMA),確定了最佳合成條件:nPEG∶nMMA=1∶1.10,反應溫度為 120 ℃,催化劑質量分數為5%,阻聚劑質量分數為2%,酯化率可達78.5%以上;然后以DBTL為催化劑,使IPDI與PAM、烷基醇封端反應,第1步最佳反應溫度60℃,反應時間2 h,第2步反應溫度65~70℃,反應時間2~2.5 h,成功制備了一類新型的反應型非離子聚氨酯表面活性劑.
(2)通過紅外光譜、核磁共振譜合成的樣品進行了結構表征,確定了其結構.對合成的反應型聚氨酯表面活性劑進行了一系列物化性能的測試.實驗證明此類表面活性劑有較高的表面活性,臨界膠束濃度CMC值可達10-5mol/L,且降低表面張力的能力隨著疏水鏈段所占比例的增加而增加.此外,該表面活性劑也具有較好的乳化能力和聚合能力,有望應用于丙烯酸酯類或其他單體O/W型乳液聚合中.
[1]張世朝,徐寶財.特種表面活性劑和功能性表面活性劑—反應型表面活性劑的研究進展[J].日用化學工業,2010,40(4):296-300.
[2]LIU L,ZHOU Q.Reactive polyoxyalkylene surfactants and their use in emulsions and dispersions:US,20090163650[P].2009-02-19.
[3]DONG Y,JIN Y.Surface activity and solubilization of a nove1 series of functional polyurethane surfactants[J].Polymer International,2007,56(1):14-21.
[4]廖波,鄭朝暉,成煦,等.聚氨酯高分子表面活性劑的研究進展[J].高分子通報,2008,27(2):27-35.
[5]ZHENG J,LUO J X,ZHOU D W.Preparation and properties of non-ionic polyurethane surfactants[J].Colloids and Surfaces A:Physicochemical and Engineering Aspects,2010,36(3):16-21.
[6]林東恩,胡守印,張逸偉.新型磺酸鹽聚氨酯表面活性劑的制備及性能[J].華南理工大學學報:自然科學版,2011,33(3):32-35.
[7]ZHANG H T,DUAN L L,CHEN L,et al.Stability and copolymerization of concentrated emulsion of styrene and butyl acrylate in the presence of polyurethane macromonomer[J].Macromonomer Journal of Applied Polymer Science,2007,103:1992-1999.
[8]朱再盛,呂廣鏞.聚氨酯型反應性乳化劑的制備與性能研究[J].華南師范大學學報:自然科學版,2003(2):76-80.
[9]陳大俊,李娜.反應型乳化劑的合成及其與丙烯酸酯乳液共聚[J].化學世界,2004(4):186-188.
[10]宋志超,胡應模,劉在美.可聚合聚氨酯型表面活性劑的制備及其性能[J].涂料工業,2006,36(12):21-23.
[11]張彪,金勇,苗青,等.反應型聚氨酯非離子表面活性劑的合成及其性能[J].中國皮革,2010,39(1):26-37.
[12]林建云,朱宏,邱志慧,等.聚氨酯類反應型高分子表面活性劑的研究進展[J].高分子通報,2012(5):89-97.
[13]LOMOLDER R,PLOMANN F,SPERER P.IPDI在聚氨酯反應中對選擇性溫度、催化過程和反應對象的影響[J].上海涂料,2001,39(5):10-14.
[14]陳志明,尹輝軍.UV固化脂族聚氨酯丙烯酸酯合成動力學研究[J].涂料工業,2004,34(4):15-17.
[15]韓冰,呂彤,梁靖宇.硼酸雙甘油葡萄糖苷月桂酸酯的合成及性能研究[J].天津工業大學學報,2012,31(2):56-58.
