染發劑中4種p-苯二胺N位取代衍生物的高效液相色譜測定法
2013-11-28 01:00:34吳光斌
分析測試學報 2013年2期
胡 陽,吳光斌,倪 輝
(集美大學 生物工程學院,福建 廈門 361021)
苯二胺類染發劑是目前應用最廣泛的染發劑原料,具有染色效果好、色調變化寬、維持時間長等諸多優點[1],也是已確認的有害物質,可經皮膚吸收,引起皮疹和使肝臟受損害,吸入粉塵可引起過敏、鼻炎、支氣管炎、發燒等不同病癥,經常染發或職業上使用含苯二胺類染發劑的人,其膀胱癌、皮膚癌和淋巴瘤患病率顯著高于常人[2-5]。p-苯二胺N位取代衍生物是染發劑中常見的染料,在化妝品中屬于嚴格禁限用物質。Jean-Luc等[6]證實p-苯二胺及其N-乙酰基和N,N-雙乙酰基取代物對人體有遺傳毒性。我國《化妝品衛生標準》(GB7916-87)[7]和《化妝品衛生規范》(2007年版)[8]對其作了嚴格的限量要求,規定該4種染料成分最大不超過6.0%。
針對種類繁多的染料成分,各國科學家一直在積極研究新的檢測方法以適應不斷增加的染發劑檢測要求,已有方法主要包括薄層色譜[9]、高效液相色譜(HPLC)[10-11]、液相色譜 -質譜(HPLCMS)[12]、氣相色譜(GC)[13]、氣相色譜 - 質譜(GC - MS)[14]、毛細管電泳[15]等。苯二胺類染料成分具有極性強、揮發性低及不穩定的特點,HPLC是較理想的檢測方法。Axel等[16]使用HPLC/DAD和ECD檢測生物樣品中的p-苯二胺及其衍生物N-乙酰基-p-苯二胺和N,N-雙乙酰基-p-苯二胺,于文蓮等[17]采用液相色譜法對染發劑中5種N-氨基N-硝基苯酚的同分異構體進行同時測定,但目前國內外針對染發劑中多種p-苯二胺N位衍生物的分析研究較少。本文建立了染發劑中N,N-雙(2-羥乙基)-p-苯二胺硫酸鹽(DHPD)、N,N-二甲基-p-苯二胺硫酸鹽(DMPD)、N,N-二乙基-p-苯二胺硫酸鹽(DEPD)、N-苯基-p-苯二胺鹽酸鹽(PPD)4種p-苯二胺N位衍生物的高效液相色譜檢測方法。
1 實驗部分
1.1 儀器與試劑
Waters 1525高效液相色譜儀,配紫外檢測器(美國Waters);PH211型pH計(意大利Hanna);KQ-250E型超聲波提取器(昆山市超聲儀器有限公司);EL104型電子天平(瑞士Mettler-Toledo,感量0.1 mg);5415D型離心機(德國Eppendorf);0.45 μm有機濾膜。
標準物質(純度≥98%):N,N-雙(2-羥乙基)-p-苯二胺硫酸鹽、N,N-二甲基-p-苯二胺硫酸鹽、N,N-二乙基-p-苯二胺硫酸鹽、N-苯基-p-苯二胺鹽酸鹽均購自日本東京化成公司。
乙腈(美國天地公司)、辛烷磺酸鈉(美國Sigma公司)均為色譜純;亞硫酸鈉、十二水合磷酸氫二鈉、磷酸二氫鉀、乙醇均為分析純。
1.2 實驗步驟
1.2.1 溶液配制 25 mmol/L磷酸鹽緩沖液體系:稱取十二水合磷酸氫二鈉0.958 g、磷酸二氫鉀3.06 g、辛烷磺酸鈉1.0 g溶于約900 mL超純水中,磷酸調節pH值至6.0,最后定容至1 000 mL,經過0.45 μm濾膜過濾;1%亞硫酸鈉溶液:稱取1.0 g亞硫酸鈉溶于約90 mL超純水中,定容至100 mL;單一標準儲備液:準確稱取62.5 mg N,N-雙(2-羥乙基)-p-苯二胺硫酸鹽、N,N-二甲基-p-苯二胺硫酸鹽、N,N-二乙基-p-苯二胺硫酸鹽、N-苯基-p-苯二胺鹽酸鹽4種標準品,各加入25 mg亞硫酸鈉,用乙醇水溶液(50%)定容至25 mL容量瓶中,混勻。該溶液的濃度為2.5 g/L,-18℃保存一周;混合標準工作溶液:分別移取適量的各單一標準儲備液,用乙醇-水溶液稀釋成濃度為0.5、1、5、50、100、200、400 mg/L的混合標準溶液。
1.2.2 樣品處理 準確稱取0.5 g樣品(精確至0.000 1 g),置于50 mL容量瓶中,加入約10 mL 50%乙醇水溶液,使樣品完全溶解,然后加入1 mL 1%的亞硫酸鈉,用50%乙醇水溶液定容。渦旋混合后超聲提取15 min,轉移至離心管以4 000 r/min離心5 min,取上清液并過0.45 μm有機濾膜后進行高效液相色譜檢測。
1.3 液相色譜條件
色譜柱:Agilent Eclipse XDB-C18柱(150 mm×4.6 mm,5 μm,美國Agilent公司);流動相:A相為25 mmol/L磷酸緩沖液體系(pH 6.0,含0.1%的辛烷磺酸鈉),B相為乙腈;梯度洗脫:0~5 min,5%~30%B,5~9 min,30%~40%B,9~13 min,40%~90%B,13~15 min,90%~5%B,15~25 min,5%B。流速:1.0 mL/min;柱溫25℃;進樣量:20 μL;檢測波長:250、280 nm。
2 結果與討論
2.1 樣品前處理
本方法采用50%乙醇水溶液超聲波提取,由于染料中間體極易氧化變質,因此在提取過程中加入1 mL 1%的亞硫酸鈉溶液作為還原劑以提高染料的回收率。樣品無需凈化,即采用提取液離心去沉淀并過濾后直接進樣,方法簡便、快速。
2.2 液相色譜條件的選擇
2.2.1 檢測波長的選擇 采用紫外分光光度計對4種目標物進行紫外區波長掃描,得到其紫外吸收光譜圖(見圖1)。由各物質的紫外吸收光譜可知,N,N-雙(2-羥乙基)-p-苯二胺硫酸鹽、N,N-二甲基-p-苯二胺硫酸鹽、N,N-二乙基-p-苯二胺硫酸鹽在250 nm有特征吸收,N-苯基-p-苯二胺鹽酸鹽在280 nm有特征吸收,因此實驗選用250 nm和280 nm作為定量檢測波長。

圖1 4種p-苯二胺N位取代衍生物標準品的紫外吸收光譜圖Fig.1 UV spectra of four N-substituted derivatives standards
2.2.2 色譜柱的選擇 相關研究表明使用C8柱分離染發劑中的染料成分效果不佳[10],因此實驗選用普通C18與普通C16柱進行分離效果的比較,結果顯示待測組分在兩種色譜柱上的保留無明顯差異,考慮到色譜柱的適用性,選擇普通C18柱對待測物進行分離。
2.2.3 流動相體系的選擇 考慮到樣品中的其它苯二胺類物質對待測4種染料成分分離的影響,根據實際樣品基質的干擾情況對流動相比例及梯度進行了優化。實驗研究了不同溶劑、pH值及離子對試劑對分離效果的影響。考察了水-甲醇、水-乙腈、磷酸鹽緩沖液體系-甲醇(用NaOH調至pH 6.0、6.5、7.0)、磷酸鹽緩沖液體系-乙腈(用NaOH調至pH 6.0、6.5、7.0)、磷酸鹽緩沖液體系-乙腈(用NaOH調至pH 6.0、6.5、7.0,含0.1%的辛烷磺酸鈉)5種流動相體系對實驗的影響,結果表明采用乙腈作為有機相較甲醇能獲得更好的峰形及保留;采用pH 6.0的流動相時,各目標物的分離效果最優;采用添加辛烷磺酸鈉的離子對試劑可以改善峰形,使4種目標物在20 min內得到良好分離,且重現性較好。因此實驗最終選擇磷酸鹽緩沖液體系-乙腈(用NaOH調至pH 6.0,含0.1%的辛烷磺酸鈉)作為最佳流動相。
由于等度洗脫難以將4種目標物與樣品中的其它染料成分有效分離。因此采用梯度洗脫,當磷酸鹽緩沖液體系-乙腈的體積比為95∶5時,可將N,N-雙(2-羥乙基)-p-苯二胺硫酸鹽與樣品中其它復雜的染料成分完全分離;緩慢增加乙腈的比例,可使較難分離的N,N-二甲基-p-苯二胺硫酸鹽和N,N-二乙基-p-苯二胺硫酸鹽得到有效分離;N-苯基-p-苯二胺鹽酸鹽的出峰時間較晚,可通過提高乙腈比例使其快速出峰。優化的流動相梯度洗脫條件如“1.3”所述。
2.2.4 柱溫的選擇 考慮到溫度對氧化型染料成分有一定的影響,因此選擇25℃作為檢測溫度,既可保證保留時間的穩定性,又可排除溫度對4種目標物的影響。
在上述優化的液相色譜條件下對標準品和實際樣品進行測定,4種染料成分標準品及樣品的液相色譜圖見圖2。由圖可見,4種目標物在20 min內得到良好分離,且峰形對稱,能達到基線分離。
2.3 標準曲線、線性范圍與檢出限
在優化實驗條件下,對不同質量濃度的4種染料標準品進行測定,結果顯示,4種染料的質量濃度在0.1~400 mg/L范圍內與其響應值呈良好的線性關系,其相關系數r為0.999 2~0.999 9。分別以S/N=3和S/N=10計算方法的檢出限與定量下限(見表1)。結果顯示,方法的檢出限為18~108 mg·kg-1,定量下限為42~320 mg·kg-1,完全滿足國家標準對染發劑的檢測要求。

圖2 4種p-苯二胺N位取代衍生物標準品(A)及染發劑樣品(B)的色譜圖Fig.2 HPLC of four dye standards(A)and four dye samples(B)peak identifications:1.DHPD;2.DMPD;3.DEPD;4.PPD

表1 4種p-苯二胺N位取代衍生物的標準曲線、線性范圍、檢出限及定量下限Table 1 Regression equations,correlation coefficients,limits of detection(LODs)and limits of quantitation(LOQs)for four dye standards
2.4 回收率與精密度
選擇染發劑5號樣品(4種p-苯二胺N位取代衍生物均未檢出),在所確定的定量下限基礎上添加3個不同水平的標準品,進行多水平的加標回收率實驗,每個加標水平平行測定6次,計算其回收率和相對標準偏差(見表2)。結果表明,4種染料成分的加標回收率為83%~115%,相對標準偏差(RSD)均小于10%,說明該檢測方法的準確度好、精密度高。
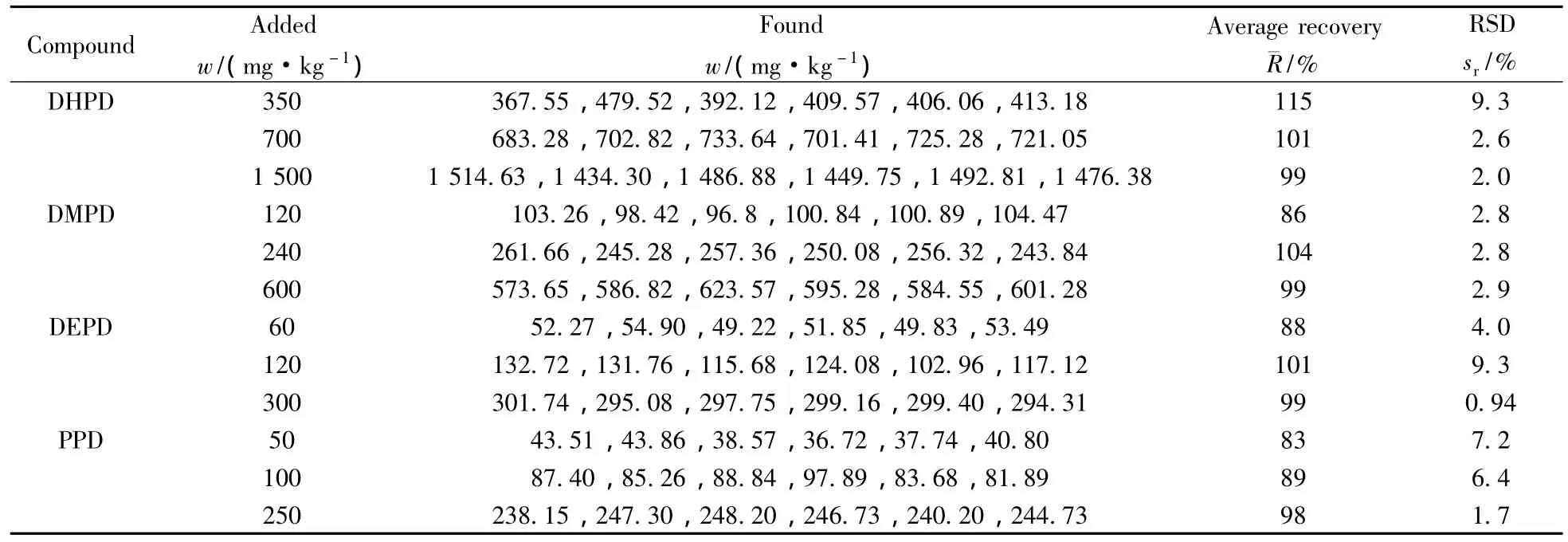
表2 4種p-苯二胺N位取代衍生物在染發劑樣品中的加標回收率及相對標準偏差(n=6)Table 2 Average recoveries and RSDs of four dye standards(n=6)
2.5 實際樣品的測定
利用本方法測定了6種市售染發劑中4種N位取代衍生物的含量(以鹽的形式計),結果見表3。所測的6種樣品中,均未檢出N,N-雙(2-羥乙基)-p-苯二胺硫酸鹽和N,N-二乙基-p-苯二胺硫酸鹽,只有6號樣品棕黑色染發劑檢出N,N-二甲基-p-苯二胺硫酸鹽;此外,除5號樣品外,其余樣品均檢出了N-苯基-p-苯二胺鹽酸鹽。

表3 市售部分染發劑中p-苯二胺N位取代衍生物含量普查結果Table 3 Detection results of some hair dyes saled in market w/(mg·kg-1)
3 結論
本文針對4種染發劑中的p-苯二胺N位衍生物建立了高效液相色譜檢測方法,該方法精密度好、回收率高、簡便、快速、準確,可用于染發劑中N,N-雙(2-羥乙基)-p-苯二胺硫酸鹽、N,N-二甲基-p-苯二胺硫酸鹽、N,N-二乙基-p-苯二胺硫酸鹽、N-苯基-p-苯二胺鹽酸鹽的測定。
[1]Brown K C,Soc J.Cosmet.Chem.,1982,33:375.
[2]Thun M J,Altekruse S F,Namboodiri M M,Calle E E.Natl.Cancer Inst.,1994,86:210 -215.
[3]Gago D M,Castelao J E,Yuan J M,Yu M C,Ross R K.Int.J.Cancer,2001,91:575-579.
[4]Andrew A S,Schned A R,Heaney J A,Karagas M R.Int.J.Cancer,2004,109:581 -586.
[5]Zhou Y G,Li Z G.J.Lab.Med.(周永貴,李真觀.環境與職業醫學),2008,25(1):100-104,107.
[6]Jean-Luc G,Mark B,Tirukalikundram K,Mel L,Gerhard J N,David K,Herve T.Mutat.Res.,2006,608:58-71.
[7]Ministry of Healthy of the People s Republic of China.Hygienic Standards for Cosmetics.GB 7916 - 1987.Beijing:China Standards Publishing House(中華人民共和國衛生部.GB 7916-1987化妝品衛生標準.北京:中國標準出版社),1987.
[8]Ministry of Healthy of the People s Republic of China.Hygienic Standards for Cosmetics,2007 ed.Beijing:Military Medical Science Press(中華人民共和國衛生部.化妝品衛生規范,2007版.北京:軍事醫學科學出版社),2007.
[9]Tamas K,Albert W G.Anal.Biochem.,2004,327:97-106.
[10]Zhu H J,Yang Y W,Zhang W Q,Zhu Y.Chin.J.Chromatogr.(朱會卷,楊艷偉,張衛強,朱英.色譜),2008,26(5):554-558.
[11]Motoko N,Kazuo M,Kauffmann J.Anal.Chim.Acta,2007,588:316-320.
[12]Yu W L,Du Z X,Wang L F,Cheng Y,Sun X,Wang Z,Li L,Zhang J.Chin.J.Anal.Chem.(于文蓮,杜振霞,王立峰,程艷,孫鑫,王崢,李蕾,張靜.分析化學),2009,37(10):129.
[13]Xue J H,Liang C,Li C.Appl.Chem.Ind.(薛金花,梁超,李程.應用化工),2011,40(2):349-352.
[14]Lai Y,Chen H X,Lin R,Wang H H,Dong Q M,Huang Z P.Chem.Res.Chin.Univ.(賴鶯,陳和秀,林睿,王鴻輝,董清木,黃宗平.高等學校化學學報),2011,32(10):2286-2292.
[15]Lu Y C,Wang H Y,Song P P,Liu S H.Chin.J.Chromatogr.(盧玉超,王海燕,宋萍萍,劉書慧.色譜),2011,29(11):1122-1127.
[16]Axel M,Brunhilde B,Klaus F.J.Chromatogr.B,2009,877:1627-1633.
[17]Yu W L,Chen W,Zhou X,Wang L F,Sun X,Wang Z,Chen H M.J.Instrum.Anal.(于文蓮,陳偉,周新,王立峰,孫鑫,王崢,陳會明.分析測試學報),2009,28(8):975-977.
