HPLC法測定注射用三磷酸胞苷二鈉的含量和有關物質
2013-12-03 03:35:48周長明韓南銀北京大學藥學院北京100091北京市藥品檢驗所北京100035
中國藥房 2013年5期
關鍵詞:質量
張 彤,周長明,紀 宏,韓南銀(1.北京大學藥學院,北京 100091;.北京市藥品檢驗所,北京100035)
三磷酸胞苷二鈉(CTP)是核苷酸類衍生物,屬于輔酶類藥物,在機體內參與磷脂類及核酸的合成和代謝,是腦磷脂合成與核酸代謝的中間產物和能量來源[1-2],對顱腦外傷后綜合征及其后遺癥有良好的治療作用。注射用CTP國家標準的含量測定方法為紙電泳法[3],該法操作煩瑣、誤差較大,且此標準中未對有關物質進行控制。由于CTP分子間存在高能磷酸鍵,注射用CTP在生產和貯存過程中很容易發生磷酸鍵斷裂,降解成一磷酸胞苷二鈉(CMP)、二磷酸胞苷二鈉(CDP)等,從而對產品的質量產生影響。
筆者建立了CTP含量測定及有關物質檢查的高效液相色譜(HPLC)法,結果表明該方法操作簡便、靈敏度高、重復性好、專屬性和耐用性較強。經方法學驗證,該方法適用于CTP的質量控制。采用本方法對12個廠家的25批市售樣品進行考察,結果表明不同廠家的產品質量存在較大差異。故本法也為廠家進一步從原輔料的選擇、處方和工藝的優化等方面提供了較強的指導意義。
1 材料
LC-20AT型HPLC儀、SPD-M20A二極管陣列檢測器、LC-solution色譜工作站(日本島津公司);AE-240天平(瑞士梅特勒-托利多公司)。
CTP對照品(蕪湖華仁科技有限公司,批號:11011909,純度:98.2%,水分:3.7%;美國Sigma公司,批號:BCBC2057,純度:95%,水分:2.1%);CDP對照品(蕪湖華仁科技有限公司,批號:11042913,純度:98.6%,水分:2.3%);CMP對照品(美國Sigma公司,批號:070MM1493V,純度:99.7%,水分:23.1%);注射用CTP市售樣品(共12個廠家25批樣品,規格:每支40 mg);四丁基溴化銨及其他試劑均為分析純;水為純化水。
2 方法與結果
2.1 色譜條件
色譜柱:Thermo Hypersil ODS C18(250 mm×4.6 mm,5 μm);流動相:磷酸鹽緩沖液(取磷酸氫二鈉17.8 g、磷酸二氫鉀6.8 g,加水900 ml溶解,用1 mol/L氫氧化鈉溶液調節pH值至7.0,加入四丁基溴化銨0.80 g,加水至1000 ml,搖勻),流速:1.0 ml/min;進樣量:20 μl;檢測波長:280 nm;柱溫:30 ℃。
2.2 溶液制備
對照品溶液:精密稱取CTP對照品適量,用流動相定量稀釋制成每1 ml中約含0.4 mg的溶液,搖勻即得。
含量測定溶液:取樣品加流動相溶解并定量稀釋制成每1 ml中約含0.4 mg的溶液,搖勻即得。
有關物質檢查溶液:取樣品3支,加流動相將內容物溶解并全量轉移至適宜的量瓶中,加流動相定量稀釋制成每1 ml中約含0.4 mg的溶液,搖勻,作為供試溶液。精密量取供試溶液5 ml,置于100 ml量瓶中,加流動相定容至刻度,搖勻,作為對照溶液。
2.3 有關物質檢測方法
2.3.1 系統適用性試驗。取對照品溶液進樣分析,理論板數按三磷酸胞苷計不低于3000,三磷酸胞苷與二磷酸胞苷峰及與其后相鄰色譜峰之間的分離度均應>2.0。含量測定按峰面積以外標法計算。有關物質記錄色譜圖至主成分保留時間的2倍,以主成分自身對照法計算有關物質的量。
2.3.2 專屬性試驗。取樣品(安徽A廠,批號:101112)適量(約相當于CTP 20 mg)3份,分別加入1 mol/L氫氧化鈉溶液1 ml、1 mol/L鹽酸1 ml、10%過氧化氫(H2O2)溶液1 ml,室溫下放置1 h后中和;另取樣品2份置于105℃烘箱中加熱破壞2 h或在紫外燈下照射1 d。分別加水定容至50 ml,取20 μl進樣分析。結果表明,本品經熱破壞后,產生的雜質峰均能與主峰基線分離,說明該色譜系統能有效檢測CTP的分(降)解產物;而本品對酸、堿、氧化和光穩定。色譜見圖1。
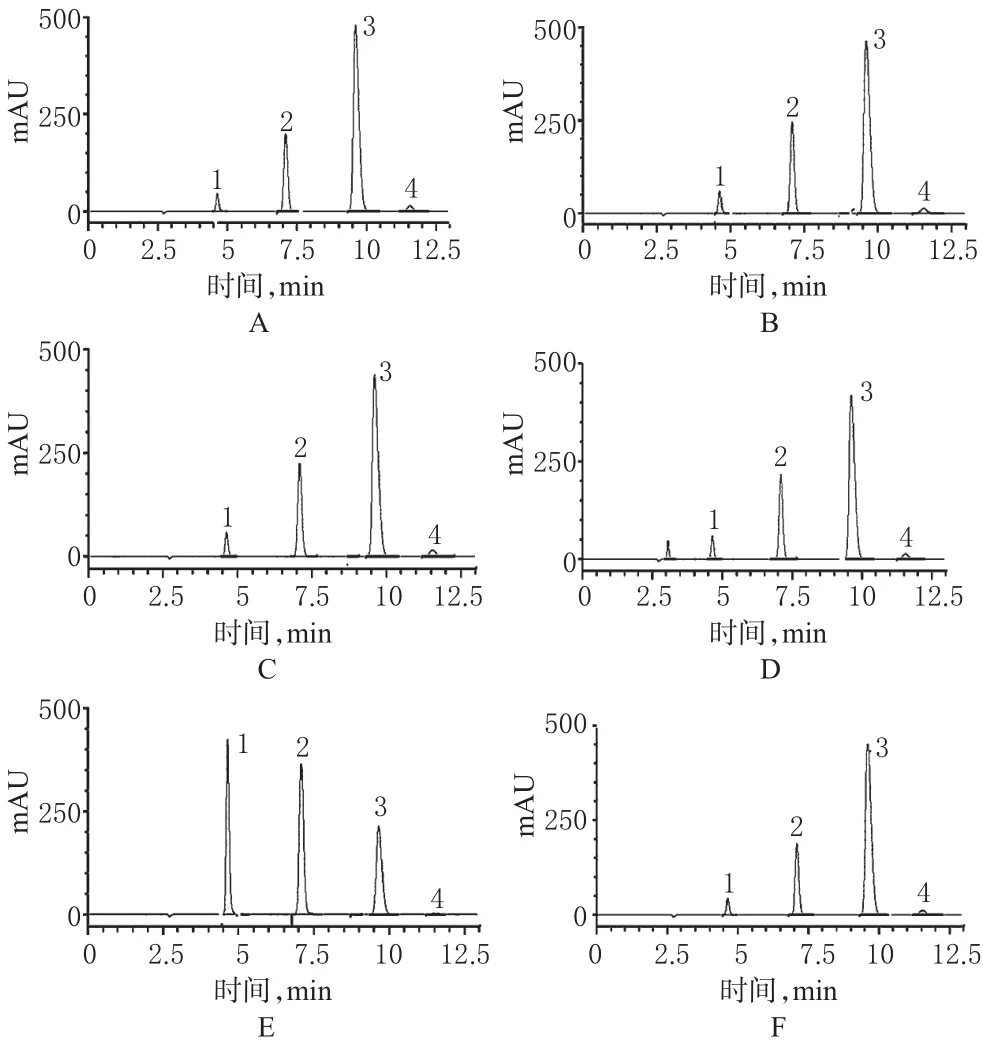
圖1 專屬性試驗高效液相色譜圖Fig 1 HPLC chromatograms of specificity tests
2.3.3 響應因子測定。用國產和進口CTP對照品、國產CDP對照品及進口CMP對照品初步考察了CDP和CMP與CTP質量濃度相同時在280 nm檢測波長處的峰面積之比。結果表明,CTP(國產)/CDP峰面積之比為0.8004,CTP(進口)/CDP之比為0.8191,CTP(國產)/CMP之比為0.6195,CTP(進口)/CMP之比為0.6340。這與CMP/CTP分子質量比值0.622和CDP/CTP分子質量比值0.811基本一致。
2.3.4 定量限。根據測試溶液質量濃度和進樣體積計算,CTP定量限(信噪比為10)為7.4 ng,CDP定量限(信噪比為10)為1.08 ng,CMP定量限(信噪比為10)為0.21 ng。
2.4 含量測定方法
2.4.1 線性關系。精密稱取進口CTP對照品約0.45 g,置于200 ml量瓶中,加水溶解并稀釋至刻度,再精密量取5 ml,分別置于10、25、50、100 ml量瓶中,加水稀釋至刻度,搖勻即得質量濃度分別為1860、930、372、186、93 mg/L的對照品溶液。精密量取上述5種溶液各20 μl進樣分析,記錄色譜圖。以質量濃度(c,mg/L)對峰面積(A)進行回歸,得回歸方程A=9.724×103c+2.156×105(r=0.9997,n=5),表明CTP檢測質量濃度線性范圍為90~1800 mg/L。
2.4.2 精密度試驗。取質量濃度約為400 mg/L的對照品溶液,連續進樣6次。結果,其峰面積的RSD=0.28%(n=6),表明方法精密度良好。
2.4.3 重復性試驗。取同一批樣品,測定6次,結果平均含量101.0%,RSD=1.4%(n=6)。
2.4.4 穩定性試驗。精密量取進口CTP對照品溶液,在室溫下經3、5、8、19、25 h放置后分別進樣。結果RSD=0.9%(n=5),表明供試品溶液在室溫下放置25 h內穩定。
2.4.5 回收率試驗。精密量取對照品溶液(約1000 mg/L)3、4、5 ml各3份,分別置于10 ml量瓶中,加處方量的輔料溶液稀釋至刻度。按含量測定方法,精密量取上述溶液各20 μl進樣分析,記錄色譜圖,計算回收率。結果,平均回收率為102.1%,RSD=1.65%(n=9),詳見表1。
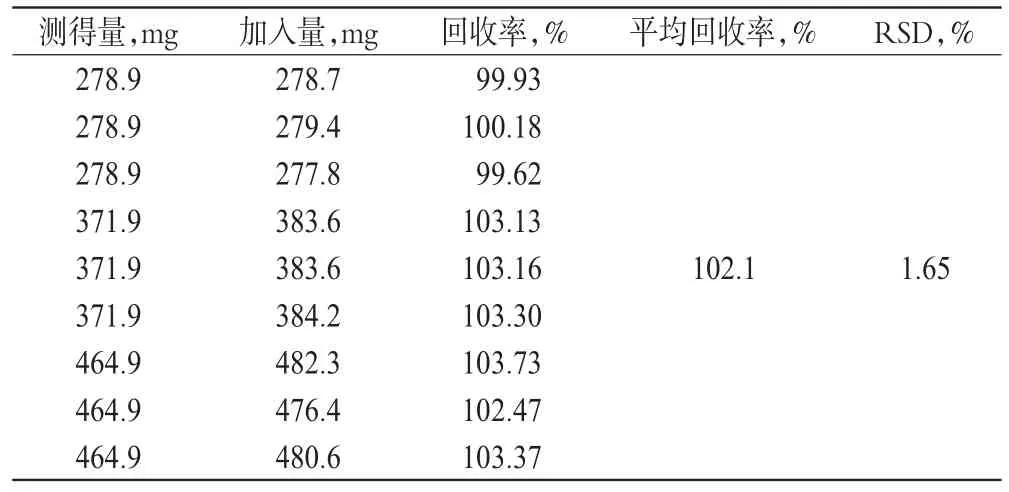
表1 回收率試驗結果(n=9)Tab 1 Results of recovery tests(n=9)
2.4.6 耐用性試驗。取同一批樣品,采用不同廠牌4種色譜柱測定,評價系統適用性相關指標。結果,CTP理論板數均>3000,其中3種色譜柱CTP與CDP分離度>2.0。
2.5 樣品測定
取各市售樣品,按“2.2”項下方法制備成含量測定樣品溶液,同時取不含CTP的輔料同法制備成空白溶液及進口CTP對照品溶液,在“2.1”項下色譜條件依法測定,記錄色譜圖見圖2(圖2為安徽A廠產品,批號:101112);含量和有關物質測定結果見表2。
3 討論
3.125 批樣品考察結果分析
根據國家藥典委員會2010-2011年度國家藥品標準提高工作的相關要求,需對注射用CTP的質量標準進行補充與提高工作。目前國內該品種共有31個批準文號,最終收集到12家企業的25批樣品。筆者根據原質量標準建立了含量和有關物質測定方法及考察了其方法學,并利用建立的方法對25批市售產品進行了質量考察和對比,結果發現注射用CTP主要市售產品間主成分含量和有關物質含量均存在顯著性差異。
3.2 流動相的考察
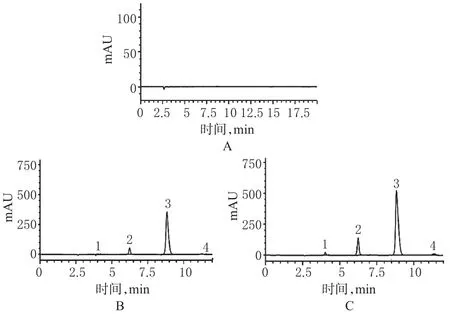
圖2 含量測定高效液相色譜圖Fig 2 HPLC chromatograms of content determination

表2 25批樣品含量和有關物質測定結果Tab 2 Results of content and related substance determination of 25 batches of samples
文獻[4-6]報道常用的流動相為水相或水相與有機相甲醇混合。筆者在前期試驗中對多種流動相進行了比較,結果發現在水相中加入甲醇會使主峰保留時間縮短。水相多采用磷酸鹽緩沖液,用氫氧化鈉或三乙胺調節pH值。pH升高會延長主峰保留時間,有利于分離,普通液相色譜柱的pH使用高限為7。在流動相中加入一定量的離子對試劑四丁基溴化銨可改善分離度。磷酸鹽濃度和離子對試劑濃度均對主峰保留時間有一定影響,通常較高的鹽濃度能縮短主峰保留時間,而增加離子對試劑濃度可改善分離度。經過對不同流動相所記錄色譜圖的綜合分析與比較(保留時間、峰形、理論板數、分離度),最終確定采用本法中的流動相。
3.3 檢測波長的確定
本試驗采用二極管陣列檢測器,所以可以方便地得到供試品的紫外圖譜。在試驗中發現,CTP在280 nm波長處有最大吸收,CMP和CDP均在279 nm波長有最大吸收,并且空白溶液在280 nm波長處無紫外吸收,因此確定280 nm為本試驗的檢測波長。
3.4 定量方法的確定
CTP含量測定的方法文獻報道很多。其中包括地標升國標(第16冊)[3]的紙電泳法。紙電泳法操作煩瑣,手動點樣準確度低,且試驗過程中干擾測定結果的因素較多,最終造成結果的重現性較差。另外也有采用HPLC法利用經典公式計算CTP的質量比(%),再結合紫外吸收系數法測定總核苷酸(即CTP、CMP和CDP及其他可能存在的核酸類成分的總量),最終計算得到CTP含量[7-10]。此方法不使用CTP對照品,主要是考慮CTP對照品一般純度不高,多含CMP和CDP等有關物質,并具有較強的引濕性,且對溫度敏感,需低溫保存。另外在計算CTP的質量比的公式中使用的0.655和0.848分別作為CMP和CDP的折算系數,但在采用不同對照品的實際測定結果顯示與此數值有差異。考慮到工藝和保存條件等的提高,對照品法是更加簡易可行的方法。
本方法也同樣適用于注射用CTP中有關物質的檢查。在保證主成分與有關物質峰的良好分離下,同時滿足為縮短試驗周期對運行時間的需要,采用自身對照法定量測定有關物質。由于CDP和CMP紫外響應高于CTP,所以實際含量低于給出的結果,但不影響不同廠家間產品的質量評價。
綜上所述,本試驗方法操作簡便、快速、專屬性強、靈敏度高、重復性和耐用性好、結果準確,與原標準比較能夠更好地控制產品的質量。本文的有關物質測定結果對有關生產企業加強原輔料質量控制、改進工藝并通過穩定性試驗考察制訂更加合理的貯藏條件和效期具有較強的指導意義。
[1]國家食品藥品監督管理局.關于修訂三磷酸胞苷二鈉制劑說明書的通知[S].2006-02-22.
[2]郭瑞芳,李英平,李玉臣,等.三磷酸胞苷二鈉對腦缺血大鼠海馬CA3區突觸體素表達的影響[J].中風與神經疾病雜志,2003,20(6):529.
[3]國家食品藥品監督管理局.化學藥品地方標準上升國家標準:16冊[S].2003:270-272.
[4]國家食品藥品監督管理局.YBH21632004三磷酸胞苷二鈉注射液[S].2004.
[5]國家食品藥品監督管理局.YBH25922005三磷酸胞苷二鈉注射液[S].2005.
[6]歷文霞,蘇云明,李國輝.HPLC法測定三磷酸胞苷二鈉氯化鈉注射液的有關物質[J].中國藥師,2010,13(4):529.
[7]國家食品藥品監督管理局.YBH10172006注射用三磷酸胞苷二鈉[S].2006.
[8]國家食品藥品監督管理局.YBH24542006注射用三磷酸胞苷二鈉[S].2006.
[9]國家食品藥品監督管理局.YBH24472006注射用三磷酸胞苷二鈉[S].2006.
[10]國家食品藥品監督管理局.YBH04792008注射用三磷酸胞苷二鈉[S].2008.
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
