高通量測序序列比對研究綜述
2014-04-29 00:44:03高靜焦雅張文廣
生命科學研究 2014年5期
高 靜 焦 雅 張文廣

摘要:高通量測序技術的飛速發展,給生物信息學帶來了新的機遇和挑戰,第二代測序序列數量多、長度短使得原來的序列分析手段不再適用。近幾年來,針對高通量測序的序列分析算法和軟件日益增多,目前已有上百種,導致選擇合適的軟件成為一個難題。對第二代測序的測序類型、序列類型以及分析算法進行了總結和歸納,對現今常用的分析軟件的序列的類型、長度以及軟件應用算法、輸入/輸出格式、特點和功能等方面做了詳細分析和比較并給出建議。分析了現今測序技術和序列分析存在的問題,預測了今后的發展方向。
關鍵詞:高通量測序:序列比對;序列作圖;序列比對工具
中圖分類號:Q-31
文獻標識碼:A
文章編號:1007-7847(2014)05-0458-07
以Roche/454焦磷酸測序(2005年)、lllumina/Solexa聚合酶合成測序(2006年)和ABI/SOLiD連接酶測序(2007年)技術為代表的第二代測序技術與Sanger測序相比,共有的突出特征是單次運行產出序列數據量大,故而又被通稱為高通量測序技術。高通量測序技術大大降低了測序的時問和成本,因而得到了廣泛應用。但隨之而來的短序列(short read)為基因數據分析帶來了新挑戰。目前對短序列一個常用的分析方法則是將已有基因組序列作為參考基因序列(reference),將短序列與參考基因序列進行序列比對,并在參考基因序列上進行定位,這個過程稱為mapping。序列比對是基岡數據分析最基本的手段,對其進行研究具有重要意義。
1 高通量測序概述
1.1 測序分類
大規模、低成本、快速的高通量測序技術被廣泛應用于生物研究的各個方面,當前主流的測序分為:1)基因組學的全基因組de novo測序、全基因組重測序、外顯子&目標區域測序、簡化基因組測序;2)轉錄組學的轉錄組測序、數字基因表達譜、小RNAs測序、降解組測序和長鏈非編碼BNA洲序;3)表觀基囚組學的全基因組Bisulfile甲基化測序、RRBS、MeDIP測序等。
1.2 測序數據類型
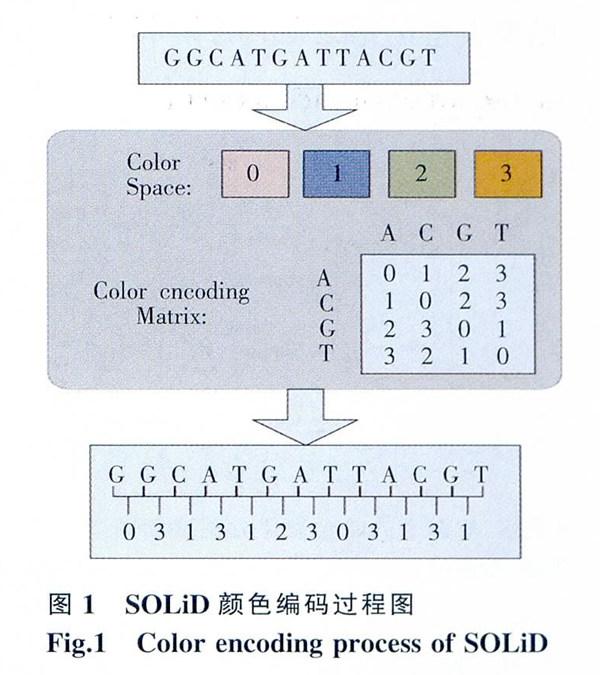
現在比較常見的測序方式有:single -read、paired-end、Mate-pair、color-space。1)Single-read即為單末端測序,在測序前先將DNA樣本進行片段化處理形成200~500 hp的片段,且將引物序列連接到DNA片段的一端,然后末端加上接頭。這種方法較簡單,但是它只有一端有拼接信息,不利于拼裝;2)Paired-end稱為雙末端測序或配對末端測序,它是指在構建待測DNA文庫時,在基因片段的兩端都加上測序引物結合位點再進行測序。雙末端測序是在片段兩端都加上接頭,在序列拼裝的時候更容易定位片段;3)Male-pair也屬于雙末端測序,與paired-end不同的是它將基因隨機打成特定為2~10 kb的片段,然后經末端修復,生物素標記和環化等實驗步驟后.再把環化后的片段打斷成400~600 bp的片段并標記測序。因為經過了環化步驟,Mate-pair比paired-end方式測得的序列片段更長,從而可以將基因序列中大量的repeat包含在內,減少拼接難度;4)Color-space read是ABI公司的SOLiD測序儀檢測出的read,SOLiD可以同時檢測兩個相鄰的堿基,并利用顏色空間的4種不同的顏色對兩個堿基編碼。對于這種編碼方式,只要知道顏色編碼對應的序列上任何一個位置的堿基類型,就可以將顏色編碼解碼為原來的堿基序列,圖l為顏色編碼過程。
2 序列比對方法
對于百萬條甚至上億條短序列在reference上的定位,傳統的動態規劃算法不能滿足我們的要求。為了加快序列比對的速度,應用啟發式算法是必然趨勢。同前,絕大多數的啟發式算法都是通過建立索引加快比對速度。建立索引就是用一個輔助的數據結構存儲待比對序列,這種數據結構能夠將序列中符合一定規則的子序列凸顯出來。常用的數據結構有哈希表和后綴樹兩種。
2.1 基于種子的哈希表索引方法
這種方法多用于數據庫搜索或read在參考序列上的mapplng,以哈希表形式建立序列的索引,這類算法的代表軟件是SSAHA,基于種子的哈希表方法具體步驟:
第一步,建立哈希表。
DNA序列由A、C、T、G四種脫氧核苷酸組成,長度為k的連續片段也就是“種子”有4K種可能將4K種子存放在哈希表中,我們用一個公式將所有種子唯一標識,將4個堿基的值f(x)轉換為二進制數據表示,只占用2 Bit的內存。
第二步,將數據庫中的序列與哈希表關聯
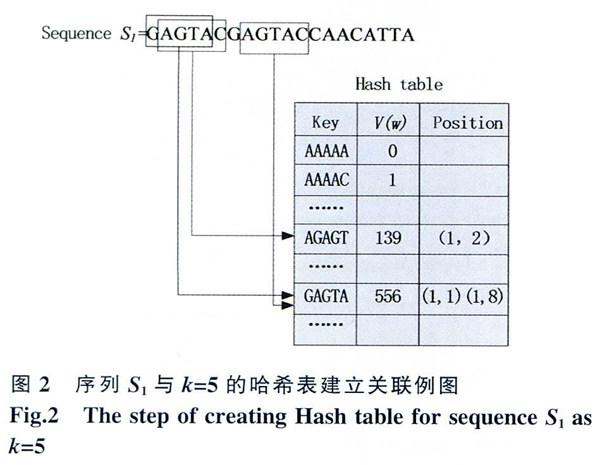
設一個含有n條DNA序列的數據庫D={S1,S2,…,Sn},將數據庫中的每條序列分解連續種子w,其長度k=5,從頭開始每次偏移一位直到序列結束,那么長度為l的序列有(l-k+1)個種子。并將每一個“種子”所在的序列,N(N=l,2,…,n)和在序列中的順序號L (L=1,2,…,l)在對應的哈希表中的“種子”以二維數組方式記錄下來(N,L)。將S1
第三步,查詢序列Q也分解成長度為k的種子并計算其標識號,在哈希表中找到數據庫中與之匹配的所有位置。
為了讓算法更高效,算法在各個方面進行了改進:1)“種子”長度,允許錯配和indel的數量等因素直接影響比對結果的準確性、敏感度、時間和空間花費等指標,所以研究者們一直致力于對“種子”形式和選擇的改進中。Blastn、Blat、SOAP、SeqMap、MAQ、SHRiMP使用不同的種子模型,如空位種子、空位種子組(同時使用多個種子模型)等;2)-些軟件如MAQ、RMAP、SeqMap、ZOOM等將reads序列建立索引,而一些軟件將參考序列數據庫建立索引;3)設置閾值F將哈希表中出現頻率低于F的“種子”刪去。
2.2 基于前/后綴樹的索引方法
后綴樹是一種重要的數據結構,AVID、MUMmer、MUMmer等序列比對算法都是基于后綴樹的這個結構,但與兩序列相似性比對時用于尋找最大公共子序列不同,read在參考序列中mapping時,是將read或其子序列在按序排列的所有后綴中一一比對找到匹配位置。算法VCAKE、SSAKE、SHARCGS、BWA-SW應用前綴樹數據結構,它將序列所有的前綴用樹的結構表示,原理與后綴樹相同。如圖3所示為序列S=GATGAC的前綴樹和前綴DAWG (DirectedAcyclic Word Graph,有向無環詞圖)。
由于后綴樹結構在比對大型序列時,空間占用比較大,Abouelhoda等改進了Manber和My-ers提出的后綴樹存儲方法,稱為增強后綴數組來提高空間利用率。Ferragina和Manzini提出的FM (full-text minute-space)-indexc是一種基于BWT (burrows-wheeler transform)的全文本壓縮索引結構,利用BWT矩陣與后綴樹之間的關系壓縮后綴數組和索引,減少內存占用率。
第一步,BWT矩陣T設序列S以“$”為結束符號,輪換列出序列S所有的后綴記為數組M,然后將數組M的每行按照字母順序排序得到BWT矩陣T;
第二步,矩陣T中第i行在矩陣M中的行號(也是矩陣T第一列字母在序列S中的序號)為數組S[i]矩陣T中最后一列的結果為BWrr數組B[i]。我們可以根據這兩個數組將序列S還原,這個算法稱為UNPERMUTE算法;
第三步,創建數組Oc (A)表示矩陣T中第一列的第一個A在第幾行,Occ(2,G)表示矩陣T的最后一列中第2行的G是此列的第幾個G。以這兩個數組為索引可以很快找到匹配點;
用這種方法比對的軟件有:Bowite、BWT-SW、BWA-SW。我們知道人類的基因組為3 Gb,那么數組Occ和S的每個值都要用4 Byte表示,3 Gb就要分別用12 GB內存存儲。在Bowtie中應用了FM-index,每隔數行建立一個索引,使得人類基因組的內存占用約為1.3 GB,完全可以在普通機器上運行。
基于這兩種數據結構的算法都有利于提高比對效率,兩者的區別在于后綴樹可以有效減少不精確匹配,并可避免比對過程中做的無用功,這個特點適用于相同物種之間相似性高的序列比對和尋找保守區。而應用哈希表數據結構的算法具有較高的敏感性,有利于發現SNPs和突變。可用于局部匹配或從大量數據巾搜索匹配點以及跨物種序列間的比對。
3 序列分析軟件比較
表1中列出民目前常用的第二代測序序列的分析軟件,對軟件分析的序列類型、序列長度、軟件特點、功能以及可輸入、輸出格式等方面進行了詳細析和比較。
選擇合適的軟件要根據軟件適用的序列類型、長度以及運用的算法、性能特點、輸入/輸出格式等項全而考慮,如表1中列出的常用軟件的各項屬性進行篩選:1)根據第2列中序列類型,如果是Spliced reads可以選擇QPALMA和TopHat軟件,(color-spaced reads可以選擇BWT -SW、BFAST;2)根據第4列中read的長度,SSAHA、BLAT用于長序列的比對;SOAP.ZOOM、SeqMap適用于short-reads的mapping;3)按照表中5和6列軟件的特點和功能,如Bowtie、BWT -SW、RMAP、MAQ、QPALMA、SHiRMP等在分析序列對,應用了質量分數,這在很大程度上提高了比對的準確性。用Bowlie和BWA在分析速度內存占用方面略優于其他軟件:4)第8列為軟件在比對時是否允許錯配,gaps以及indels,這也影響比對結果。例如,MAQ在比對Illumina paired-end reads時允許gaps這樣可以加大paired-end reads拼接的成功率。BWA、BFAST、SHiRMP、PASS等在進行比對時允許一定或不定數量的gaps可以提高比對的敏感度,降低假SNP的出現率,有利于發現真正的突變位點;5)最后,根據軟件的輸入格式是否符合待比對序列的格式、輸出格式是否又符合下一步分析要求的格式進行軟件選擇。
此外,在分析數據的時候將兩個或多個軟件結合使用,例如在處理spliced reads時,可以使用Bowtie或BWT建立索引文件,再將索引文件使用TopHat、SoapSplice等軟件進行splice junction檢測。
4 結論
本文對國內外第二代測序技術以及數據的分析算法進行了總結和分析,對目前流行的序列分析軟件進行詳細歸納和比較并給出了參考建議,有利于從整體上把握測序技術和序列分析的發展狀況.為今后序列分析和研究提供參考。
通過上述研究發現雖然基因分析軟件已有很多且解決了一定的問題,但仍然存在困難:1)由于序列中大量repeat(重復片段)的存在,很多短reads作為repeat的一部分,不能對其準確定位,增大了拼裝難度; 2)當前大多硬件環境較低,計算時間以小時或天為單位,嚴重限制了研究進展;3)比對結果的準確率和下一步分析的可用性方面有待提高。
針對上述問題,第三代單分子納米孔測序技術目的是在第二代測序技術的基礎上提高測序的準確性、增長read的讀長。雖然其技術還不成熟,但必將成為未來幾年的主流技術。序列比對方面,現有越來越多的軟件提供并行處理方式,所以算法并行化以及云計算技術的應用將會成為序列分析的重要發展方向。
