17-Demethoxy-reblastatin聯合順鉑對三陰性乳腺癌細胞增殖和凋亡的影響
2014-07-07 15:29:33趙素容董清清蔣琛琛吳成柱
安徽醫藥 2014年3期
趙 晴,劉 浩,趙素容,張 配,董清清,蔣琛琛,吳成柱
(蚌埠醫學院藥學系,安徽省生化藥物工程技術研究中心,安徽 蚌埠 233030)
17-Demethoxy-reblastatin聯合順鉑對三陰性乳腺癌細胞增殖和凋亡的影響
趙 晴,劉 浩,趙素容,張 配,董清清,蔣琛琛,吳成柱
(蚌埠醫學院藥學系,安徽省生化藥物工程技術研究中心,安徽 蚌埠 233030)
目的 觀察17-Demethoxy-reblastatin(17-DR)聯合順鉑(cisplatin,DDP)對三陰性乳腺癌 (triple-negative breast cancer,TNBC)細胞株MDA-MB-231增殖和凋亡的影響及其相關的分子機制。方法 用MTT法、集落克隆形成法檢測藥物對細胞增殖抑制的影響,流式細胞術檢測藥物作用后細胞的凋亡情況。結果 不同濃度17-DR或DDP對細胞增殖具有抑制作用,且二者聯用時抑制作用增強。50 μmol·L-117-DR、8 μmol·L-1DDP及二者聯用刺激24 h誘導細胞的凋亡率依次為14.7%、20.1%、45.2%。聯合用藥組Caspase-3的激活增強以及下調RIP1蛋白的表達。結論 17-DR作為熱休克蛋白90(heat shock protein 90,Hsp90)抑制劑能夠增強DDP對MDA-MB-231細胞增殖抑制和誘導凋亡的作用,其機制可能與下調Hsp90的顧客蛋白RIP1的表達相關。
Hsp90抑制劑;17-Demethoxy-reblastatin;順鉑;三陰性乳腺癌;凋亡
2006年 Bryan等首次明確提出雌激素受體(ER)、孕激素受體(PR)和人表皮生長因子受體-2 (HER-2)均為陰性的乳腺癌為三陰性乳腺癌(triple-negative breast cancer,TNBC),這一定義已被廣泛接受[1-2],已成為 乳腺癌 研究 的 新熱 點 之 一。TNBC類患者約占全部乳腺癌的15%,發病年齡早,多見于絕經前女性,且早期易發生局部復發和遠處轉移,增殖指數高,無病生存時間和總生存時間均較短,目前國內外仍缺乏針對該特殊類型乳腺癌的規范化治療指南[3]。TNBC的ER、PR和 HER-2表達缺失,所以無法針對這些靶點進行治療。而且TNBC具有典型的分子遺傳學特征,如BRCA1、EGFR、Eaveolin-1等基因分子突變或過度表達,這些異常有望成為今后 TNBC靶向治療的靶點。Synta制藥公司研發的一種熱休克蛋白 90(heat shock protein 90,Hsp90)抑制劑Ganetespib(STA-9090)正處于治療 TNBC的Ⅱ期臨床研究[4]。
17-Demethoxy-reblastatin(17-DR)是在首個確證的Hsp90天然抑制劑格爾德霉素(geldanamycin,GA)的基礎上,通過產 GA菌株的突變菌株采用微生物發酵技術提取分離得到,研究已經明確其為Hsp90 N末端抑制劑[5](圖1)。Hsp90是分子伴侶中重要的一類,在癌基因蛋白的折疊、穩定及成熟等過程中發揮著重要的作用。Hsp90在細胞內以同型二聚體形式存在,每個亞單位都由3個結構域構成:N-端的 ATP酶域,與顧客蛋白結合的中間域及包含蛋白與蛋白間相互作用和二聚化修飾的 C-端域[6]。由于 Hsp90 N末端有一個高度保守的 ATP結合位點,Hsp90發揮作用的機制包括 ATP結合到其N端結構域及隨后的 ATP水解,Hsp90的伴侶活性需要ATP在這一位點的結合和水解。在 ATP結合的狀態下,Hsp90經過了構型的改變而變成成熟的復合體,這對發揮它對顧客蛋白的正確折疊及穩定作用必不可少。ATP水解產生 ADP使這些顧客蛋白的釋放變得容易隨后它們通過泛素蛋白酶體途徑而降解[7]。
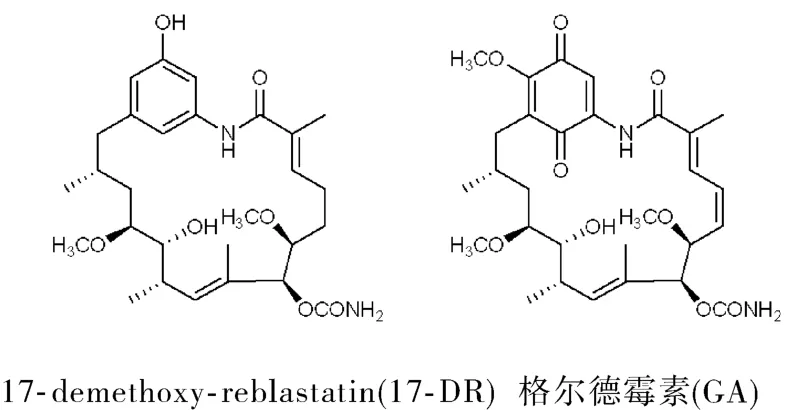
圖1 17-DR和格爾德霉素的結構
Hsp90抑制劑通過抑制 Hsp90,可間接的調節癌基因蛋白從而發揮抗腫瘤作用,且 Hsp90抑制劑對癌細胞的作用相比正常細胞有顯著的選擇性[8]。臨床前研究顯示惡性乳腺癌組織相對于正常乳腺組織其細胞質、細胞核 Hsp90染色呈顯著的高水平,同時 Hsp90 mRNA也呈高水平,另外高水平的Hsp90與乳腺癌患者的差預后也密切相關。
雖然TNBC對包括順鉑(cisplatin,DDP)在內的鉑類等化療藥物較敏感但容易產生耐藥[9],此外,研究顯示 DDP對 Hsp90有很高的親和力,DDP能結合到Hsp90的 C末端結構域并且抑制 Hsp90在C末端形成同源二聚體,導致 Hsp90蛋白的構象改變,并能特異性抑制 Hsp90介導的分子伴侶活性[10-11]。而17-DR以及 GA類化合物是結合到Hsp90的N末端結構域發揮其抑制作用,17-DR與DDP通過不同的機制抑制Hsp90介導的分子伴侶活性,理論上二者的聯合應用將能夠更大程度的抑制Hsp90。因此本研究以TNBC細胞株 MDA-MB-231細胞為研究對象,觀察了 Hsp90抑制劑 17-DR聯合DDP對乳腺癌細胞增殖及凋亡的影響,探究17-DR能否增強MDA-MB-231細胞對順鉑的敏感性,并初步探討其機制。
1 材料與方法
1.1 細胞株 人乳腺癌 MDA-MB-231細胞,購于中科院上海細胞庫。
1.2 主要試劑 DMEM培養基、胰蛋白酶:Gibco公司;胎牛血清:浙江四季青公司;MTT:Sigma公司;碘化丙啶(propidium iodide,PI):沃宏公司;兔抗人 Caspase-3抗體:abcam公司;兔抗人 BCL-2、BAX抗體:中杉金橋公司;兔抗人RIP1抗體及鼠抗人 β-actin抗體:Santa Cruz公司;17-DR:韓國生命工學研究院(KRIBB)的Dr.Hong,Young-Soo惠贈;DDP:山東齊魯制藥廠。
1.3 細胞培養 乳腺癌細胞 MDA-MB-231采用DMEM培養液,含 10%胎牛血清,3.7 g·L-1碳酸氫鈉,1×105IU·L-1青霉素,100 mg·L-1鏈霉素,37℃、飽和濕度、5%CO2培養箱中培養。
1.4 MTT法檢測細胞存活率 取處于對數生長期細胞,用0.25%胰蛋白酶消化制成單細胞懸液,調整細胞密度為 5×107·L-1,接種于 96孔細胞培養板,實驗設調零組(培養液、MTT、DMSO)、空白對照組(細胞、培養液、MTT、DMSO)和藥物處理組(細胞、培養液、藥物、MTT、DMSO),每個組設5個復孔。每孔中加100 μL含5 000個細胞的培養液,在37℃、飽和濕度、5%CO2培養箱中培養使細胞貼壁。培養24 h之后,棄去每孔培養液,換新鮮含 10%胎
牛血清培養液,加藥后 24、48、72 h(藥物作用終點時間)終止培養,終止培養4 h,每孔加MTT 15 μL,4 h后棄去上清液,每孔加入 DMSO 100 μL,37℃孵育10 min,酶標儀上振蕩5 min,用酶標儀檢測波長570 nm處吸光度(A)值,計算細胞存活率/%:細胞存活率(%)=(實驗組A值-調零組A值)/(對照組A 值-調零組 A值)×100%。以上實驗重復3次。
1.5 集落克隆實驗 用含 10%胎牛血清的 DMEM高糖培養基調整細胞濃度后,分別以每孔 1×104個細胞接種于6孔板中,24 h后吸棄培養液,5 μmol· L-117-DR、0.8 μmol·L-1DDP及兩者合用處理細胞,并設對照組,置CO2孵箱中培養 8 d,觀察到細胞集落形成后,去除培養基,預冷 PBS洗滌兩遍,20%甲醇于-20℃固定10 min,結晶紫染色,ddH2O緩慢洗去染色液,室溫下干燥,拍照觀察集落的形成情況。
1.6 流式細胞儀檢測細胞凋亡 將對數生長期的細胞制成單細胞懸液接種于 6孔細胞培養板,每孔1×105個細胞,培養24 h細胞貼壁后按照實驗設計加入藥物處理,繼續培養24 h后收集細胞至 10 mL離心管,2 000 r·min-1,離心 10 min,去上清。用PBS洗滌一次并轉移到 5 mL離心管,2 000 r· min-1,離心5~10 min,去上清,各管分別加入冰上預冷的75%乙醇1 mL固定,置于4℃冰箱過夜。第二天取出各管,2 000 r·min-1,離心5 min,去上清。各管分別加3 mL PBS重懸,2 000 r·min-1,離心5 min,去上清。各管分別加入 600 μL的 PI染液染色,室溫,避光反應30 min,上流式細胞儀(Becton Dicknson C6)進行檢測,以上實驗重復 3次。檢測具有亞G1期DNA含量的細胞比例,代表凋亡細胞數。
1.7 Western blot檢測蛋白表達 收集穩定表達細胞用細胞裂解液(總體積200 mL:100 mmol·L-1Tris-HCl(pH 7.4)20 mL、1mol·L-1NaCl 28 mL、100 mmol·L-1CaCl21 mL、100 mmol·L-1MgCl221 mL、15 mmol·L-1NaN340 mL、Triton X-100 2 mL、用前加入蛋白酶抑制劑 12 mmol·L-1Leupeptin、1 mmol·L-1PMSF各2 mL·L-1)冰上裂解 30 min,提取細胞總蛋白,BCA蛋白定量法(參照試劑盒說明書操作)測各組蛋白濃度,用細胞裂解液將各組蛋白稀釋至等濃度,與 2×上樣緩沖液 1∶1混合,100℃煮沸5 min變性。取蛋白40 μg每組,10%或15%SDS-PAGE凝膠電泳(70 V,30 min,90 V,90 min);轉膜以恒流50 mA,轉移 2.5 h至 PVDF膜;5%脫脂牛奶室溫封閉2 h(或4℃過夜);PBS洗凈,孵一抗(1∶1 000),室溫孵育2 h(或4℃過夜);TPBS洗滌3次,PBS洗滌1次;孵二抗(1∶2 000),室溫孵育2 h;TPBS洗滌3次,PBS洗滌1次;ECL發光試劑盒暗室發光、顯影、定影,Bio-Rad凝膠成像系統獲取圖像。
1.8 統計學方法 采用SPSS16.0軟件。實驗數據主要為計量數據,均行正態性檢驗,以均數 ±標準差(±s)表示。多組間比較為單因素方差分析,各組間比較用雙側Dunnett t檢驗。當 P<0.05時,差異有統計學意義。
2 結果
2.1 17-DR增強 DDP對乳腺癌細胞 MDA-MB-231增殖的抑制作用 實驗中使用不同濃度的 17-DR、DDP單用及 DDP聯合 17-DR刺激細胞,使用MTT法檢測細胞存活率。結果表明(圖2),不同濃度17-DR和 DDP單獨處理對細胞均有一定的增殖抑制作用,17-DR和DDP作用細胞 72h的IC50分別為74.9、7.0 μmol·L-1。50 μmol·L-117-DR單用作用于細胞24、48、72 h的存活率分別為86.8%、76.2%、49.9%,8 μmol·L-1DDP單用作用于細胞24、48、72 h的存活率分別為79.7%、54.5%、37.3%,50 μmol·L-117-DR與不同濃度的 DDP聯用對MDA-MB-231細胞的增殖抑制較 DDP單用時作用增強。在DDP濃度為8 μmol·L-1和17-DR濃度為50 μmol·L-1兩者合用時,細胞24、48、72 h的存活率分別為46.3%、28.4%、25.4%,與單用組在相對應濃度及時間點數據比較均有顯著性差異(P <0.05)。
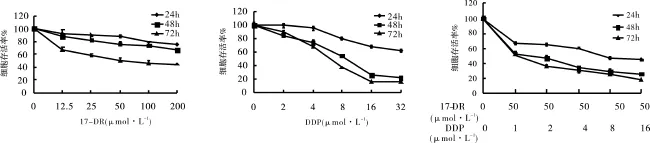
圖2 17-DR、DDP及17-DR與 DDP聯用組對細胞的增殖抑制作用
2.2 17-DR增強 DDP對乳腺癌細胞 MDA-MB-231集落克隆形成的抑制作用 為進一步觀察 17-DR對MDA-MB-231細胞增殖抑制作用及其對DDP抑制細胞增殖作用的影響,實驗中根據 MTT實驗結果使用低于IC50的 5 μmol·L-117-DR和/或 0.8 μmol·L-1DDP刺激細胞,用集落克隆形成實驗檢測藥物對細胞增殖抑制作用的影響。結果表明,與對照組相比17-DR和 DDP單獨處理組均能抑制細胞的集落克隆形成,并且二者聯用時抑制作用增強(圖3)。
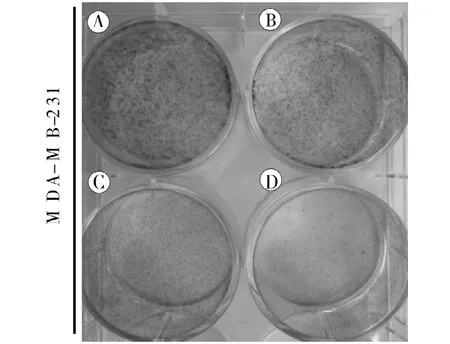
圖3 17-DR和/或DDP對乳腺癌MDA-MB-231細胞集落克隆形成的抑制
2.3 17-DR增強DDP誘導乳腺癌細胞MDA-MB-231凋亡的作用 實驗中使用 50 μmol·L-117-DR、8 μmol·L-1DDP及 50 μmol·L-117-DR與8 μmol·L-1DDP聯用刺激誘導細胞24 h,隨后對實驗中不同因素處理的細胞按照操作進行 PI染色,通過流式細胞儀分析,觀察兩藥單用或合用誘導細胞凋亡的作用,各組的凋亡率用對應Sub-G1期細胞所占的比例表示(圖 4)。50 μmol·L-117-DR、8 μmol·L-1DDP處理組誘導細胞的凋亡率分別為14.7%、20.1%與陰性對照組 1.1%比較有顯著性差異(P<0.05)。用50 μmol·L-117-DR和8 μmol ·L-1DDP聯用組相同條件處理,誘導 MDA-MB-231細胞的凋亡率增加到45.2%與對照組比較有顯著性差異(P<0.05)。
2.4 17-DR增強 DDP誘導 MDA-MB-231細胞Caspase-3蛋白的激活 實驗結果表明:17-DR刺激細胞后,并未出現明顯的 Caspase-3的激活,但 17-DR對 DDP誘導 MDA-MB-231細胞 Caspase-3的激活具有增強作用,Caspase-3蛋白裂解片段愈加明顯(圖5A)。
2.5 17-DR和/或 DDP處理對細胞 BAX、BCL-2及 RIP1蛋白表達的影響 細胞經各處理組作用24 h后,Western blot結果顯示,促凋亡蛋白 BAX的表達為聯合用藥組高于單獨用藥組,抑凋亡蛋白 BCL-2的表達為聯合用藥組低于單獨用藥組。同時 17-DR可下調細胞RIP1的表達,DDP也能使 RIP1表達下調且二者合用組RIP1下調更明顯(圖5B)。細胞依次經control組,17-DR組(50 μmol·L-1),DDP組(8 μmol·L-1),DDP(8 μmol·L-1)與17-DR(50 μmol·L-1)聯用組處理。圖5C中 a、b、c依次為BAX、BCL-2和 RIP1蛋白的表達灰度值統計結果。*P<0.05 vs control組。

圖4 17-DR增強DDP誘導MDA-MB-231細胞凋亡的敏感性注:A.control組;B、C、D:17-DR組(50 μmol·L-1),DDP組(8 μmol·L-1),DDP(8 μmol·L-1)與 17-DR(50 μmol· L-1)聯用組。
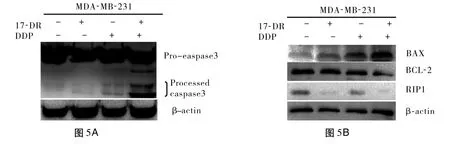
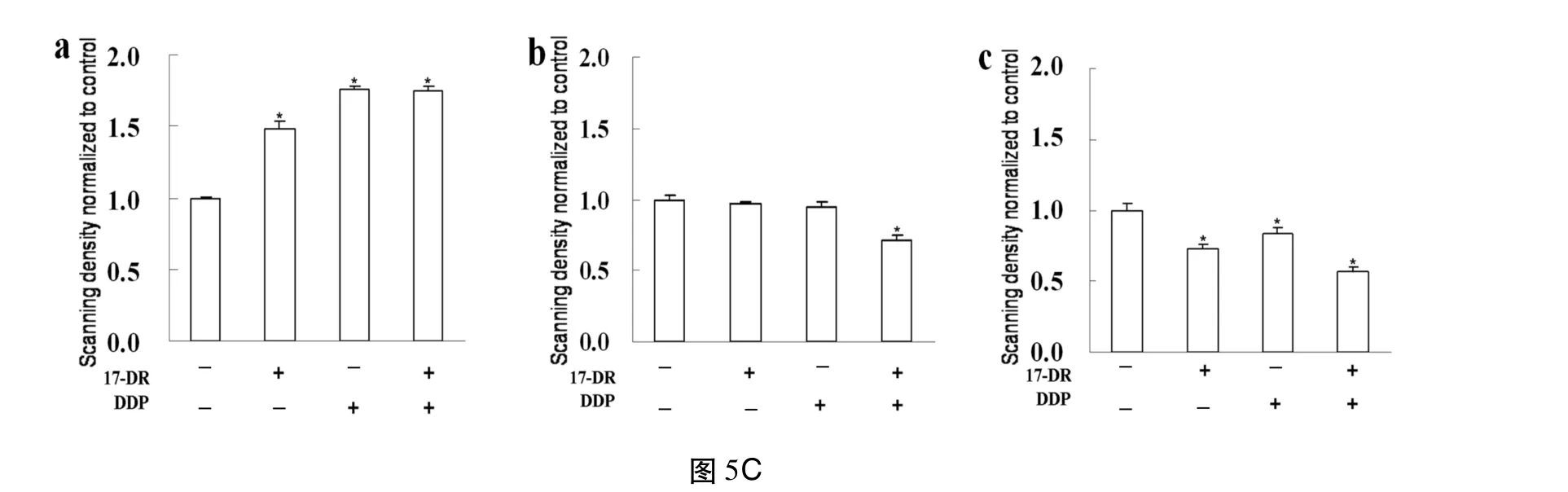
圖5 MDA-MB-231細胞Caspase-3、BAX、BCL-2和RIP1蛋白的表達情況及BAX、BCL-2和RIP1蛋白的表達灰度值統計
3 討論
目前已有多種 Hsp90抑制劑進入治療乳腺癌臨床試驗階段[12],另有多個 Hsp90抑制劑在臨床前乳腺癌研究中也顯示出較好的作用,通過高通量篩選發現的一種新型的口服 Hsp90抑制劑 PF-4942847,在多個TNBC細胞株及異種移植模型的體外和體內試驗中,顯示了顯著的抗腫瘤活性[13]。Hsp90抑制劑用于治療包括TNBC在內的乳腺癌具有良好的應用前景。GA能夠與ATP競爭結合Hsp90 N末端而使 Hsp90的顧客蛋白不能受到處于關閉狀態的 Hsp90二聚體的保護,繼而通過泛素蛋白酶體途徑降解。雖然,GA具有較好的抗癌活性,但由于GA水溶性差,肝毒性強,使其在臨床應用上受到了很大限制。因此,開發水溶性更好、毒副作用更低的GA衍生物成了 GA開發研究的主要方向。微生物因代謝產物豐富而成為了目前研究Hsp90抑制劑的主要來源,17-DR通過組合生物合成技術針對引發肝毒性結構,即苯醌結構進行結構改造,制備得到的具有非苯醌結構的GA衍生物[5]。腫瘤細胞中大多數與逃避細胞凋亡及對生長抑制信號失去敏感性有關的蛋白的穩定性依賴于Hsp90,Hsp90可以調控多種信號通路網絡上的蛋白的折疊,在腫瘤的發展和惡化中扮演著重要的角色。以 Hsp90為靶點的大多數藥物比選擇性的致癌基因通路抑制劑具有更大的優勢,因此以整個信號通路網絡而不是單一的信號通路為靶向的藥物設計,可能會解決這一問題。
DDP是具有細胞毒性的細胞周期非特異性藥物,是目前最常用的抗腫瘤藥物之一,它物美價廉,治療效果良好,臨床中主要以靜脈注射和腹腔用藥的治療效果最為明顯,其主要作用部位在DNA的嘌呤和嘧啶堿基,可抑制癌細胞的DNA復制過程,并損傷其細胞膜上結構,有較強的廣譜抗癌作用,骨髓抑制作用較輕,在多種實體瘤的治療中 DDP是有效的抗腫瘤藥,但是能夠導致腎毒性。研究表明Hsp90抑制劑 PU-H71和 DDP二者聯用下,能夠極大增強對 PU-H71抵抗癌細胞株對誘導的凋亡的敏感性[14]。但是DDP又是一種由濃度決定其治療效果的抗腫瘤藥物,在短時間內,每次給藥的濃度越高,效果也越好,但隨著用藥量的增加,出現的不良反應亦相應的增加。因此,本實驗使用低濃度 DDP單用或與17-DR合用誘導細胞24 h為主,亦是基于以上原因,從而使實驗研究與臨床的治療聯系更加密切。
RIP1為絲氨酸/蘇氨酸蛋白激酶,是一種重要的細胞信號轉導分子。它不僅參與了細胞的凋亡,還參與了細胞存活、細胞程序性壞死等多種信號的轉導并在其中起關鍵作用,其功能受泛素化、鋅指蛋白及Hsp等的調節。
本課題組Huang等[15]的研究已經表明在乳腺癌中己糖激酶抑制劑2-脫氧-D-葡萄糖(2-DG)可以通過下調RIP1增加TRAIL誘導的凋亡,從而達到抑制 TNBC細胞的作用,顯示RIP1在乳腺癌中起重要的作用。
RIP1蛋白已經明確為 Hsp90的顧客蛋白,RIP1在多個組織內表達,是各種膜整合細胞內外應力信號必需的傳感蛋白,它除了能激活 NF-κB對抗凋亡外,還可以誘導細胞死亡,包括凋亡和非Caspase依賴的壞死[16]。Hsp90可以與 RIP1結合,是 RIP1發揮作用的輔助分子。Hsp90的功能抑制劑 GA會導致RIP1的降解,隨后抑制了 TNF誘導的 NF-κB激活,使細胞對TNF誘導的凋亡更加敏感。用 GA的類似物17-DMAG處理乳腺癌細胞,細胞內 RIP1的表達下降,細胞對 TRAIL誘導的細胞凋亡更加敏感,表明Hsp90在維持 RIP1的穩定性和可溶性方面發揮著重要作用[17-18]。實驗結果顯示在 MDAMB-231細胞中RIP1蛋白高表達,17-DR能下調細胞RIP1蛋白的表達,可能通過減少NF-κB激活的途徑來促進細胞凋亡。
本研究結果顯示 17-DR能增強 DDP對 MDAMB-231細胞的增殖抑制作用,同時17-DR增強DDP對 MDA-MB-231細胞集落克隆形成的抑制作用,17-DR也增強DDP誘導MDA-MB-231細胞凋亡的作用。其機制可能是通過下調Hsp90的顧客蛋白RIP1,以及17-DR增強DDP誘導MDA-MB-231細胞Caspase-3蛋白的激活,導致抑凋亡蛋白BCL-2的表達下調,同時使促凋亡蛋白BAX的表達增加相關。Caspase-3是細胞凋亡的主要執行者,其激活的增強也表明17-DR和 DDP在相應作用濃度下二者聯用是有效的。另外 BAX過度表達時細胞凋亡增多,而BCL-2過度表達時,其產物可與 BAX結合,從而抑制了凋亡[19]。RIP1作為細胞死亡受體介導的凋亡途徑中重要的上游信號蛋白,對其表達水平的改變在一定程度上參與了細胞凋亡的調控。17-DR增強 DDP抗腫瘤作用機制比較復雜,是否還有其它作用機制參與,有待進一步研究。且本實驗中僅選用了體外培養的細胞株進行觀察,進一步的體內外藥效及機制研究有待深入。
[1] Hyslop T,Michael Y,Avery T,et al.Population and target considerations for triple-negative breast cancer clinical trials[J].Biomark Med,2013,7(1):11-21.
[2] Den Hollander P,Savage MI,Brown PH.Targeted Therapy for Breast Cancer Prevention[J].Front Oncol,2013,3:250.
[3] Loi S,Pommey S,Haibe-Kains B,et al.CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer[J].Proc Natl Acad Sci USA,2013,110(27):11091-11096.
[4] Shastry M,Yardley DA.Updates in the treatment of basal/triplenegative breast cancer[J].Curr Opin Obstet Gynecol,2013,25 (1):40-48.
[5] Wu CZ,Jang JH,Ahn JS,et al.New geldanamycin analogs from Streptomyces hygroscopicus[J].J Microbiol Biotechnol,2012,22(11):1478-1481.
[6] Jackson SE.Hsp90:Structure and Function[J].Top Curr Chem,2013,328:155-240.
[7] Li J,Buchner J.Structure,Function and Regulation of the Hsp90 Machinery[J].Biomed J,2013,36(3):106-117.
[8] Kim LS,Kim JH.Heat Shock Protein as Molecular Targets for Breast Cancer Therapeutics[J].J Breast Cancer,2011,14(3):167-174.
[9] Eckstein N,Haas B.Platinum-based chemotherapy in triple negative breast cancer[J].Dtsch Med Wochenschr,2012,137(7):333 -336.
[10]孟 帥,山廣志,李卓榮.Hsp90抑制劑的研究進展[J].中國抗生素雜志,2011,36(4):241-248.
[11]Wang G,Ye Y,Yang X,et al.Expression-based in silico screening of candidate therapeutic compounds for lung adenocarcinoma [J].PLoS One,2011,6(1):e14573.
[12]Travers J,Sharp S,Workman P.HSP90 inhibition:two-pronged exploitation of cancer dependencies[J].Drug Discov Today,2012,17(5/6):242-252.
[13]Mehta PP,Whalen P,Baxi SM,et al.Effective targeting of triplenegative breast cancer cells by PF-4942847,a novel oral inhibitor of Hsp 90[J].Clin Cancer Res,2011,17(16):5432-5442.
[14]Gallerne C,Prola A,Lemaire C,et al.Hsp90 inhibition by PU-H71 induces apoptosis through endoplasmic reticulum stress and mitochondrial pathway in cancer cells and overcomes the resistance conferred by Bcl-2[J].Biochim Biophys Acta,2013,1833(6):1356-1366.
[15]Huang YY,Liu H,Li Y,et al.Down-regulation of RIP1 by 2-deoxy-D-glucose sensitizes breast cancer cells to TRAIL-induced apoptosis[J].Eur J Pharmacol,2013,705(1/3):26-34.
[16]Bai L,Xu S,Chen W,et al.Blocking NF-κB and Akt by Hsp90 inhibition sensitizes Smac mimetic compound 3-induced extrinsic apoptosis pathway and results in synergistic cancer cell death[J].Apoptosis,2011,16(1):45-54.
[17]Carmen P,Ana IP,Abelardo LR.Down-regulation of RIP expression by 17-dimethylaminoethylamino-17-demethoxygeldanamycin promotes TRAIL-induced apoptosis in breast tumor cells[J].Cancer Lett,2010,287(2):207-215.
[18]李 莉,閆 言.受體相互作用蛋白激酶RIP1在細胞信號傳導途徑中作用的研究進展[J].基礎醫學與臨床,2011,31 (10):1165-1167.
[19]王上偉,劉 寧,韓坤元.斑蝥酸鈉對胃癌 SGC-7901細胞增殖的影響及其機制的研究[J].安徽醫藥,2013,17(4):558-560.
《安徽醫藥》2012年影響因子為1.045,在全國117家醫藥綜合類期刊中名列第5位,創歷史新高(數據由2013年版中國科技期刊引證報告擴刊版提供)。
《安徽醫藥》編輯部
Effects of 17-Demethoxy-reblastatin combined with cisplatin on proliferation and apoptosis of triple-negative breast cancer cell line
ZHAO Qing,LIU Hao,ZHAO Su-rong,et al
(Faculty of Pharmacy,Bengbu Medical College;Anhui Engineering Technology Research Center of Biochemical Pharmaceuticals,Bengbu,Anhui 233030,China)
Objective To investigate the effects of 17-Demethoxy-reblastatin(17-DR)combined with cisplatin(DDP)on the proliferation and apoptosis of triple negative breast cancer MDA-MB-231 cell line,and to explore the molecular mechanisms underlying.Methods The inhibition of cell proliferation was examined using MTT assay and colony formation assay,and the apoptosis analyzed using flow cytometry.Results Treatment with 17-DR or DDP inhibited the proliferation of the cell,and the cell viability in the combined treatment group was lower than 17-DR or DDP group alone.The apoptosis rates were 14.7%,20.1%,and 45.2%following treatment with 50 μmol·L-117-DR,8μmol·L-1DDP and combined treatment for 24 h,respectively.The combined treatment enhanced the activition of Caspase-3,and down-regulated the expression of RIP1.Conclusions As a heat shock protein 90(Hsp90)inhibitor,17-DR,could enhance DDP-induced cell death.The underlying mechanism may relate to down-regulating the expression of Hsp90 client protein RIP1.
heat shock protein 90 inhibitor;17-Demethoxy-reblastatin;cisplatin;triple-negative breast cancer;apoptosis
10.3969/j.issn.1009-6469.2014.03.007
國家自然科學基金面上項目(No 81372899);國家自然科學基金青年科學基金項目(No 81302671);安徽省青年科學基金項目(No 1408085QH162);蚌埠醫學院科技發展基金重點項目(No BYKF12A03)
趙 晴,男,碩士研究生
劉 浩,男,教授,碩士生導師,研究方向:生化藥理,E-mail:liuhao6886@foxmail.com
2013-09-20,
2013-11-19)
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中老年保健(2022年6期)2022-08-19 01:41:48
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
中國生殖健康(2019年2期)2019-08-23 08:11:42
中國生殖健康(2019年6期)2019-01-06 09:20:12
中國生殖健康(2019年5期)2019-01-06 09:16:40
祝您健康(2018年5期)2018-05-16 17:10:16
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
