500~700 μm直徑雙層空心微球制備技術研究
2014-08-07 06:25:16劉一楊劉梅芳陳素芬張占文
原子能科學技術 2014年4期
關鍵詞:實驗
劉一楊,李 潔,李 婧,蘇 琳,劉梅芳,陳素芬,張占文
(中國工程物理研究院 激光聚變研究中心,四川 綿陽 621900)
在慣性約束聚變(ICF)物理實驗中,燃料容器使用較多的是由低Z元素材料組成的三層塑料空心微球,由外至內主要由燒蝕層(CH層)、阻氣層(PVA層)、推進層(PS層)組成,制備方法有多種[1-3]。三層塑料空心微球通常直徑要求為200~400 μm,其中用于制備三層空心微球的PS-PVA雙層塑料空心微球直徑要求也在這一范圍。但隨著我國高功率激光器的飛速發展,ICF物理實驗對PS-PVA雙層空心微球的尺寸要求逐漸增大,雙層塑料微球直徑需達到500~700 μm,甚至更高,而原有的雙層空心微球制備技術難以制備直徑為450 μm以上的雙層空心微球。
因此,為滿足未來的靶丸需求,本文在原有雙層空心微球制備技術的基礎上進行技術改進,以制備出滿足要求的較大直徑的雙層空心微球。這些改進包括攪拌槳葉輪優化技術和PS微球的臭氧化表面改性技術及重新進行體系密度匹配調整。
1 實驗
1.1 實驗試劑與儀器
PVA(相對分子質量124 000~186 000,百靈威公司);鄰苯二甲酸二丁酯(DBP,分析純,天津市科密歐化學試劑有限公司);癸二酸二-(2-乙基己基)酯(DOS,化學純,上海國藥集團);鄰苯二甲酸二辛酯(DOP,分析純,上海國藥集團)。
攪拌器(歐洲之星強力控制型,IKA公司)。
1.2 實驗步驟
將制備好的PS微球(內部含水的PS球殼)進行臭氧化表面改性處理,清洗后轉移至一定濃度的PVA溶液中充分浸泡。而后移除體系中過剩的PVA溶液,將殘余的PS-PVA體系分批次加入至DOP中進行分散,再將分散后的體系一并轉移至預先配制好的油相溶劑中,持續攪拌,待PVA液膜固化后用甲苯清洗去除表面附著的連續相溶劑,即得到PS-PVA雙層空心微球。實驗流程如圖1所示。
1.3 復合液滴在流場中的形變分析
對于在一般剪切流場中運動的液滴,其形變程度決定了液滴是否能破裂為兩個或更多的小液滴。在本工作的體系中,分散相是一固體核心被液膜包覆形成的復合液滴,不可能像單一液滴一樣在發生較大形變下依然存活。當形變開始時,PS球面上某處的PVA液膜將開始變薄,形變達到一定程度后PVA液膜從PS球表面被油相溶劑剪切剝離,暴露出PS球殼,進而被油相溶劑溶解,這是通常難以獲得更大直徑PS-PVA雙層空心微球的最主要原因。因此,分析PS-PVA復合液滴在流場中的形變情況有助于確定制備大直徑雙層空心微球的方法。
Davis等[4]在文獻[5-6]的基礎上,研究了內部包含固體核心的復合液滴在攪拌流場中運動時的形變情況。設G為剪切速率;a和b(a>b)分別為無形變復合液滴和固體球的直徑;μ′和μ0分別為液滴和外流體的黏度;σ為液滴和外流體間的界面張力;α、λ、k均為無量綱參數。則:

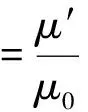
(1)
形變函數f0可表示為:
(2)
其中:ε為無量綱形變常數;K和ν為關于λ和α的方程。

(3)

圖1 PS-PVA雙層空心微球制備流程
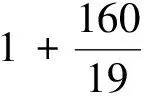
(4)
可見,液滴形變隨λ和k(k>0)的增大而降低。在本工作的體系中,油相的黏度μ0已非常低,很難找到可替代的更低黏度的溶劑,α為固體核心與復合液滴的直徑比,在雙層球直徑及PVA厚度要求固定時是常數,而ICF實驗對塑料微球的表面質量要求較高,不能引入表面活性物質進行表面張力的改善。因此可用來改善大直徑雙層空心微球存活能力的因素,包括提高λ和降低G。提高λ最直接的方式是提高PVA液膜的初始濃度,但初始濃度高會增加形成目標尺寸的PS固體核心-PVA液膜復合液滴的難度。實驗中,當PVA溶液的濃度超過5%(質量分數,余同)時,常溫下形成獨立的包覆體系就已相當困難。圖2示出不同濃度下PVA溶液μ′隨溫度的變化情況,可看出,隨溫度的升高,μ′迅速下降,而λ隨溫度的升高而增大(圖3),這是由于油相與PVA溶液的黏度隨溫度的變化趨勢不一致所導致的,因此增大λ較好的方法是增大PVA濃度的同時提高固化溫度。
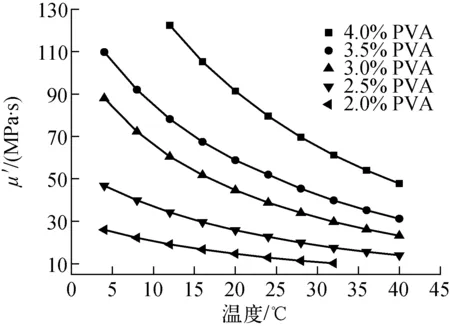
圖2 PVA溶液μ′-溫度曲線
由于攪拌槳和容器尺寸固定,降低攪拌速率可降低G,但由于在容器中遠離攪拌中心的區域流場速度非常緩慢,導致液滴出現運動遲緩、聚集與粘連的情況,這使大部分液滴最終不能固化形成獨立微球,因此需調整攪拌槳結構以優化容器內的流場。此外通過臭氧化改性PS微球,提高PVA液膜與PS殼壁之間的作用強度,進而使得PVA液膜不易被溶劑從PS微球表面剝離,提高了復合液滴的抗剪切能力。
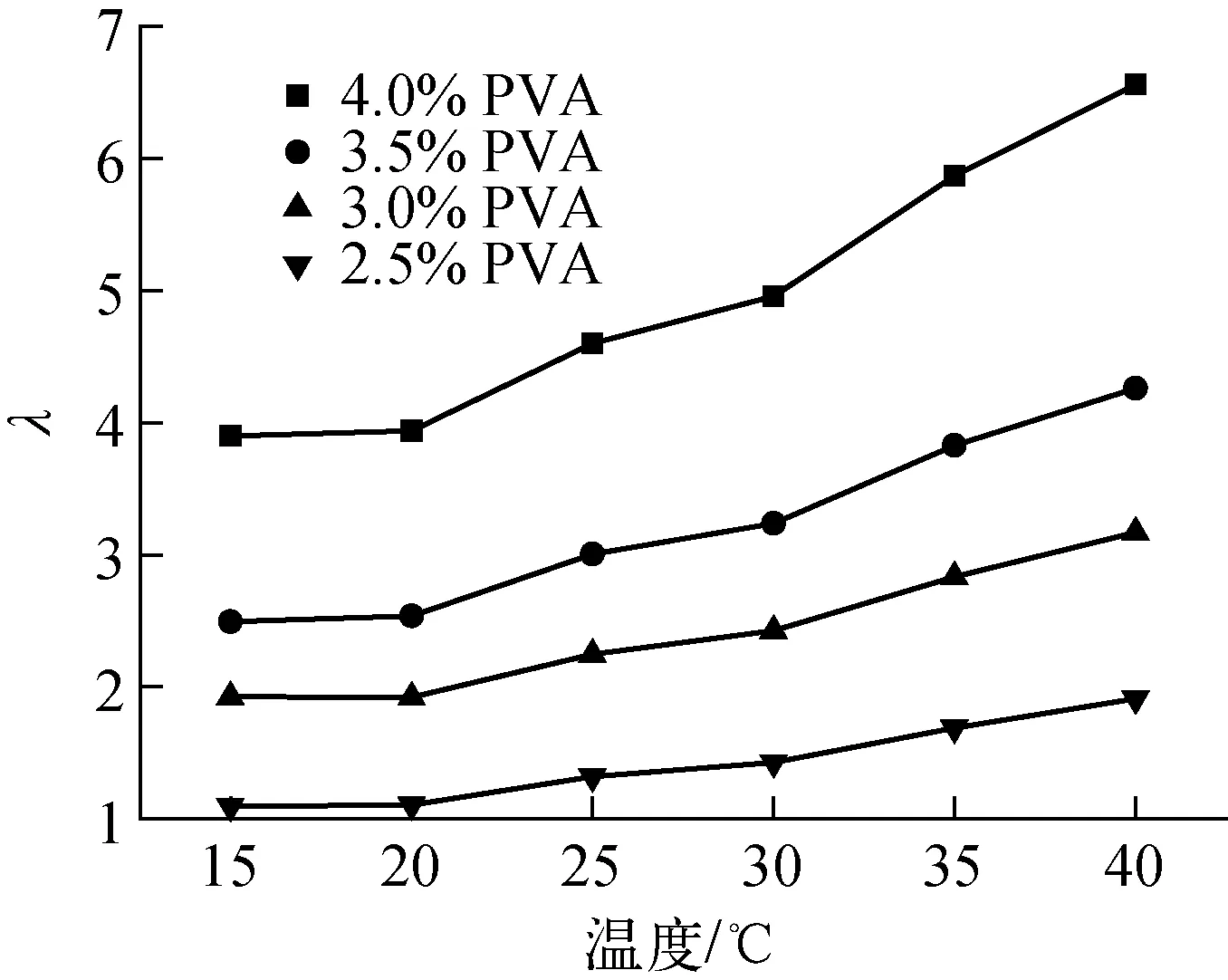
圖3 PVA溶液與連續相溶劑λ-溫度曲線
2 結果與討論
2.1 PS空心微球的臭氧化改性
使用臭氧化表面改性技術對PS微球表面進行了處理。文獻[7]的研究結果表明,PS薄膜在酸性條件下會發生臭氧化交聯反應。一方面表面生成大量極性基團,使得PS憎水的表面轉變為親水表面,對水接觸角降低;另一方面在PS表面形成交聯覆蓋層,提高了PS球殼抵抗油相溶劑侵蝕的能力。
對于固液界面,液體對固體的潤濕性F可表示為:
F=γLVcosθ=γSV-γSL
(5)
粘附功W為:
W=γLV+γSV-γSL
(6)
由式(5)和(6)可得:
W=γLV(cosθ+1)
(7)
實際情況中,氣相是被液相L′所取代,則式(7)可變換為:
W=γLL′(cosθ+1)
(8)
其中:γLV為液氣界面張力;γSV為固氣界面張力;γSL為固液界面張力;θ為固液接觸角。
因此,固液接觸角的減小(圖4)使粘附功W增加,即PVA液膜在PS表面被剝離時需外界做更多的功,這意味著在流場中PS-PVA的復合液滴將能抵抗更強的剪切。納米劃痕測試表明,臭氧化處理后,固態PS薄膜與PVA薄膜間的作用強度增大約40%,這使已成形的PS-PVA雙層空心微球不易出現“脫層”現象。

圖4 處理前、后接觸角的變化
本文通過上述技術制備了500~700 μm大直徑雙層空心微球,圖5示出微球的可見光顯微鏡照片和X光照片。可看出,照片中存在PVA壁厚均勻性不好的問題。

圖5 雙層空心微球可見光顯微鏡照片(a)和X光照片(b)
2.2 攪拌槳葉輪結構優化
實驗中通常使用的三葉片攪拌槳葉片結構與尺寸均固定。在使用這種攪拌結構制備大直徑雙層空心微球的過程中,經常遇到的問題為:PS固體核心-PVA液膜組成的復合液滴在槳葉附近(圖6中Ⅰ區域)隨流體運動時,速率較大,剪切較強,PVA液膜易從PS表面剝離;在容器底部(圖6中Ⅱ區域)運動時,由于流體運動速率較小,液滴運動速率較小,甚至在重力作用下很難隨流體向上運動,當液滴在底部大量堆積時會產生粘連;隨著PS球內核心水的遷移擴散,復合液滴密度逐漸降低導致液滴上浮至油相的氣液界面并沿器壁(圖6中Ⅲ區域)運動,此時邊緣流體速率亦非常小,上浮的復合液滴相互易產生粘連。
圖6為優化前、后容器內流體的流動速度分布(圓柱形容器沿軸線的截面)。可看出,在保證攪拌結構主體區域流體速度大致相等的情況下(攪拌葉片轉速相等),優化后的攪拌結構在底端和器壁周圍流體的速度分布有了明顯的改善(模擬顯示底端流體速率提高了3~6倍),而葉輪附近流場的運動速率基本保持不變,微球運動至容器底部時能隨流體盤旋向上運動形成循環,實驗采用優化的攪拌結構能明顯改善微球的存活率,且攪拌葉輪的轉速可進一步降低,使微球在攪拌葉輪附近受到的剪切減小,從而提高了整個流場范圍內K的最小值,使更大直徑的復合液滴得以存活。
2.3 液滴與溶劑初始密度匹配對PVA壁厚均勻性的影響
PVA壁厚均勻性是PS-PVA雙層塑料微球的一項重要參數,受PS固體核心密度、PVA液膜密度以及復合液滴體系與油相密度之間的匹配關系等影響較大。本實驗考察在PVA濃度4.5%,固化溫度37 ℃和20 ℃條件下,外連續相密度與PS-PVA復合液滴初始密度差對PVA厚度均勻性的影響。為降低微球之間由于PVA膜層厚度差異所帶來的影響,定義一PVA厚度不均勻性參量H=(最大壁厚-最小壁厚)/平均壁厚。由于實驗中采用的PS微球與PVA溶液的密度差在0.004 g/cm3附近很小幅度地變化,為簡化實驗,不考慮PS微球與PVA液膜密度差帶來的影響。圖7為37 ℃和20 ℃條件下連續相溶劑密度(ρc)與PS-PVA復合液滴密度(ρd)差ρc-ρd的平均值與平均PVA厚度不均勻性的關系。

圖6 優化前、后攪拌葉輪周圍流體及速度分布情況
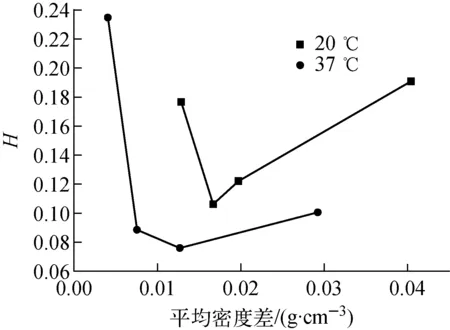
圖7 平均壁厚不均勻性-平均密度差曲線
由圖7可看出,隨平均密度差的增大,壁厚不均勻性先變小后變大。因為隨時間的增長,PVA液膜較PS核心中的水更快擴散至溶劑體系,這導致PS-PVA液滴體系整體密度增加、PVA黏度增大及PVA液膜中的水向溶劑擴散速率降低。隨著PS核心水的擴散,當其向PVA中的擴散與PVA液膜中的水向外連續相的擴散平衡時,液滴體系的密度開始降低。在實驗中還觀察到,當PS核心內開始出現明顯的空泡時,PVA液膜流動性通常已變得非常差,可認為此時PVA在溶劑中已基本不具有形變能力。由實驗結果可做出如下推斷:最初,復合液滴體系密度略低于連續相,隨水的擴散,液滴體系密度接近或超過連續相,ρc-ρd過小時,由于在溶劑底部的流體運動緩慢使液滴體系自轉緩慢,而當ρc-ρd過大時液滴可能在PVA液膜仍有較好的流動性時浮于液面,這均導致最終PVA層厚度不均勻性變大。
實驗考察了作為固體核心的PS微球不圓度與固化后PS-PVA雙層微球不圓度的關聯情況,不圓度D=(最大直徑-最小直徑)/平均直徑。圖8中,DPVA和DPS分別為固化后測得的PS-PVA雙層空心微球和內部PS球的不圓度。由于實驗采用的PS微球壁厚大于5 μm,可將PS微球視為剛性球體,不隨實驗過程發生形變,因此最終測得的DPS數據代表其初始的球形偏離情況。
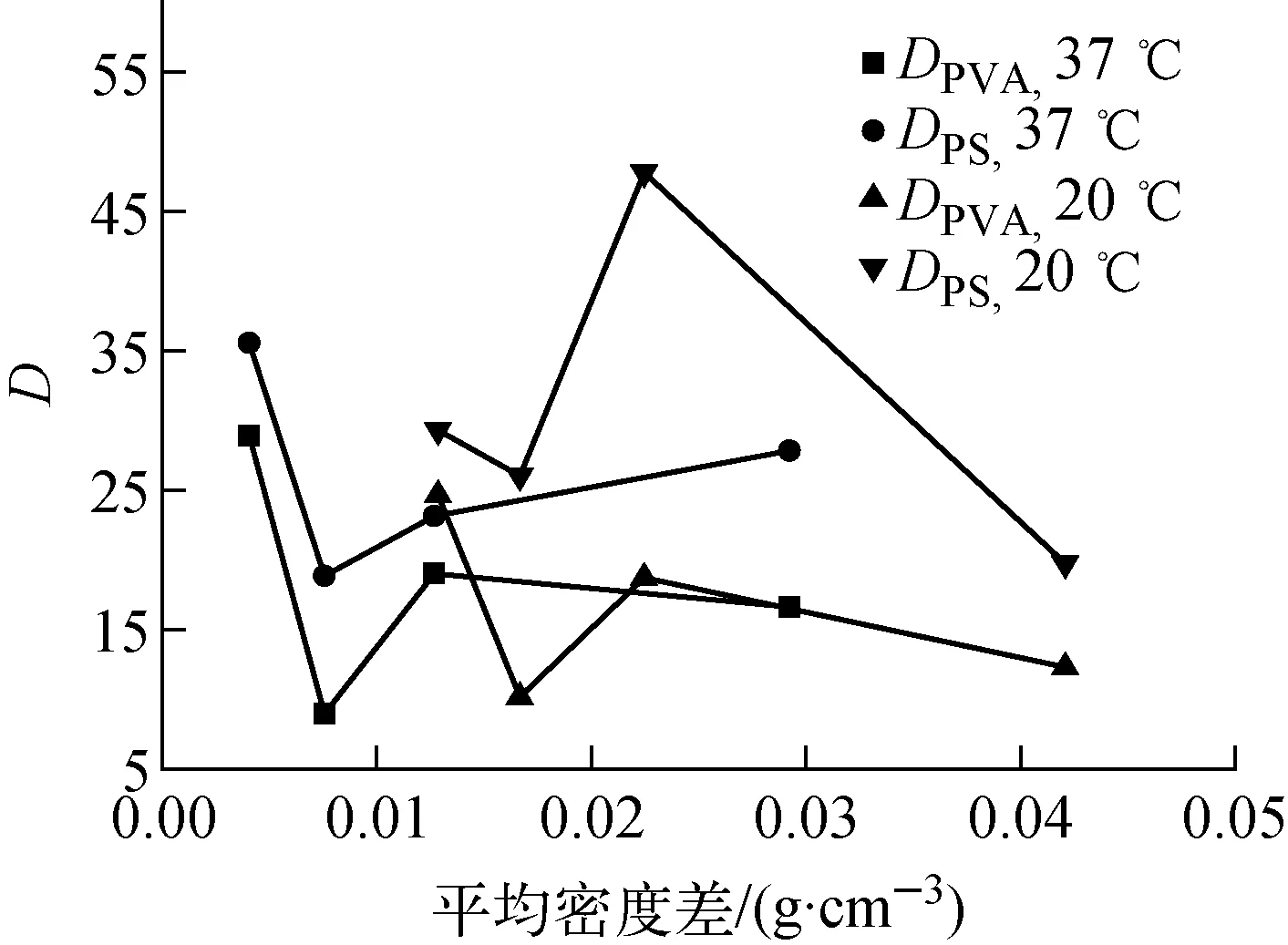
圖8 20 ℃及37 ℃下PS和PS-PVA球的不圓度
由圖8可發現,PS固體核心的不圓度曲線與復合液滴固化后PS-PVA雙層微球不圓度曲線變化規律相近,這說明PVA液膜固化過程對于PS基底的形狀特性存在明顯的復制作用。但由于PVA液膜在表面張力的作用下仍會趨于球形,因此PS微球不圓度增大必然導致PVA涂層均勻性變差。對比圖7與8可發現,在密度差較小的范圍內PS-PVA微球不圓度與PVA厚度不均勻性變化規律較相近,PS固體核心的形狀因素可能干擾了密度匹配帶來的影響;當密度差較大時兩者偏離較大,雖然PS固體核心與PS-PVA微球的不圓度較小,但獲得的PVA涂層厚度均勻性較差,顯然此時密度匹配的影響超過了PS固體核心不圓度的影響,這可能是由于過大的密度差導致復合液滴在PVA液膜仍具有較好流動性時就懸浮在液面上,液滴自轉受阻,且受流場控制其運動不再是無規的,這導致PS固體核心與PVA液膜之間的密度差變得不可忽略。因此圖7中的曲線并不能完全體現密度匹配所帶來的影響,獲得較真實的密度匹配因素對于PVA厚度均勻性的影響,還需更精確的實驗,保證PS固體核心球形度、壁厚均勻性、直徑分布都非常好。同時受X射線測量精度的影響,PVA的厚度分布也需控制在較窄范圍內才能實現,這將在后續的工作中進行報道。
3 結論
采用優化的攪拌器葉輪結構和對PS微球進行臭氧化表面改性的方法,降低了PS固體核心-PVA液膜復合液滴在外連續相溶劑中的剪切強度,提高了PS核心-PVA液膜間的粘附強度,制備了直徑500~700 μm、PVA涂層厚度2~6 μm、厚度均勻性較好的大直徑雙層空心微球。
參考文獻:
[1] 張林,涂海燕,唐永健,等. ICF靶PVA-PS雙層空心微球的研制[J]. 高分子材料科學與工程,1997,13(S1):136-138.
ZHANG Lin, TU Haiyan, TANG Yongjian, et al. Preparation of PS-PVA double-layered hollow microspheres for ICF target[J]. Polymer Materials Science & Engineering, 1997, 13(S1): 136-138(in Chinese).
[2] 李波,張林,張占文,等. 乳液微封裝技術制備聚苯乙烯空心微球[J]. 中國核科技報告,2003(1):81-94.
LI Bo, ZHANG Lin, ZHANG Zhanwen, et al. Preparation of polystyrene hollow microsphere by emulsion microencapsulation technology[J]. China Nuclear Science and Technology Report, 2003(1): 81-94(in Chinese).
[3] 游丹,李波,張林,等. 界面縮聚法PS-PVA雙層聚合物空心微球的研制[J]. 強激光與粒子束,2000,12(4):463-466.
YOU Dan, LI Bo, ZHANG Lin, et al. Preparation of PS-PVA double-layered hollow microshells by interface poly-condensation technique[J]. High Power Laser and Particle Beams, 2000, 12(4): 463-466(in Chinese).
[4] DAVIS A M J, BRENNER H. Emulsions containing a third solid internal phase[J]. Journal of the Engineering Mechanics Division, 1981, 107(3): 609-621.
[5] TAYLOR G I. The deformation of emulsions in definable fields of flow[J]. Proceedings of the Royal Society of London Series A, 1934, 146: 501-523.
[6] COX R G. The deformation of a drop in a general time-dependent fluid flow[J]. Journal of Fluid Mechanics, 1969, 37(3): 601-623.
[7] 劉一楊,闞秋斌,魏勝,等. 臭氧化法表面改性聚苯乙烯薄膜研究[J]. 強激光與粒子束,2011,23(1):111-114.
LIU Yiyang, KAN Qiubin, WEI Sheng, et al. The study of surface modification of PS films by ozone[J]. High Power Laser and Particle Beams, 2011, 23(1): 111-114(in Chinese).
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
