脂多糖加速飲食所致非酒精性脂肪肝小鼠的炎癥和氧化應激
2014-08-15 09:06:04史國兵張敬一
實用藥物與臨床 2014年10期
張 威,史國兵,張敬一
0 引言
非酒精性脂肪肝(Nonalcoholic fatty liver disease,NAFLD)是一種最常見的慢性肝臟疾病,在成人和兒童中都有較高的發病率[1],非酒精性脂肪肝炎(Non-alcoholic steatohepatitis,NASH)的特點是具有顯著的脂肪病變、炎癥和進行性的肝纖維化,最終導致肝硬化甚至是肝癌的發生[2]。腸道細菌、細菌毒素以及內毒素誘導的前炎癥因子(如腫瘤壞死因子-α能激起脂肪性肝炎的壞死性炎癥病變和肝纖維化[3-4],并且有臨床研究表明,NASH患者腸道中的菌群含量顯著高于正常受試者[5]。另外,有臨床以及實驗室研究證明抗菌藥物能夠改善肝功能[6-7]。提示腸道來源的細菌性內毒素可能會誘發NASH的發生,因此,本研究旨在探討脂多糖在飲食誘導的NASH模型中的作用。
1 材料和方法
1.1 實驗動物及分組 本實驗選用8~10周齡雄性C57BL/6小鼠,體重(20±2) g,購自中國醫科大學動物實驗中心,動物合格證號:SCXK(遼)2008-0005。實驗動物常規飼養于沈陽軍區總醫院清潔級動物房,12 h光照和黑夜循環,溫度(22±2)℃,濕度50%~60%。MCD飼料的配方參考國外文獻[8]。MCS飼料配方[9],即MCD對照飼料,是在MCD飼料配方基礎上,加上膽堿2 g/kg、蛋氨酸3 g/kg。MCD及MCS飼料均由江蘇南通特洛菲飼料有限公司加工制作,為清潔級飼料,4 ℃低溫保存。動物分為3組,MCS或MCD飼料喂養2周,每周2次腹腔注射生理鹽水或脂多糖(LPS),組別如下:MCS+Saline組給予MCS+生理鹽水腹腔注射,MCD+Saline組給予MCD+生理鹽水腹腔注射,MCD+LPS組給予MCD+1 mg/kg LPS腹腔注射(購自Sigma)。
1.2 標本處理及組織病理學檢測 各組小鼠末次腹腔注射6 h后乙醚麻醉,摘取一側眼球后取血,3 000 rpm離心后留上清凍存備用。脫頸處死動物取部分肝小葉用10%福爾馬林固定,石蠟包埋,進行HE染色和Sirius Red染色,另取部分肝臟于-80 ℃凍存待測。
1.3 血清ALT、AST和TNF-α含量測定 ALT、AST按照試劑盒要求采用全自動生化分析儀進行測定,檢測TNF-α的ELISA試劑盒購自R&D公司,按試劑盒的操作規程對上述指標進行檢測。
1.4 肝臟TBARS含量測定 TBARS(硫代巴比妥酸反應物)能夠反映氧化應激水平,按照LabAssayTM的TBARS試劑盒的操作規程檢測各組小鼠肝臟中的TBARS水平。
2 結果
2.1 脂多糖加速NASH小鼠的肝臟炎性發生 肝臟組織HE染色可見:MCS+Saline組小鼠肝組織肝竇清晰可見,肝索排列整齊,形態結構均正常;MCD+Saline組小鼠肝組織充滿大量大小不一的脂肪空泡,肝細胞結構紊亂呈氣球樣變,有部分炎性細胞浸潤明顯,呈灶狀分布,炎性細胞以單核細胞、淋巴細胞為主;MCD+LPS組同樣也呈現出脂肪肝炎的特點,但炎性細胞浸潤較多,局部呈現壞死狀病變。見圖1。
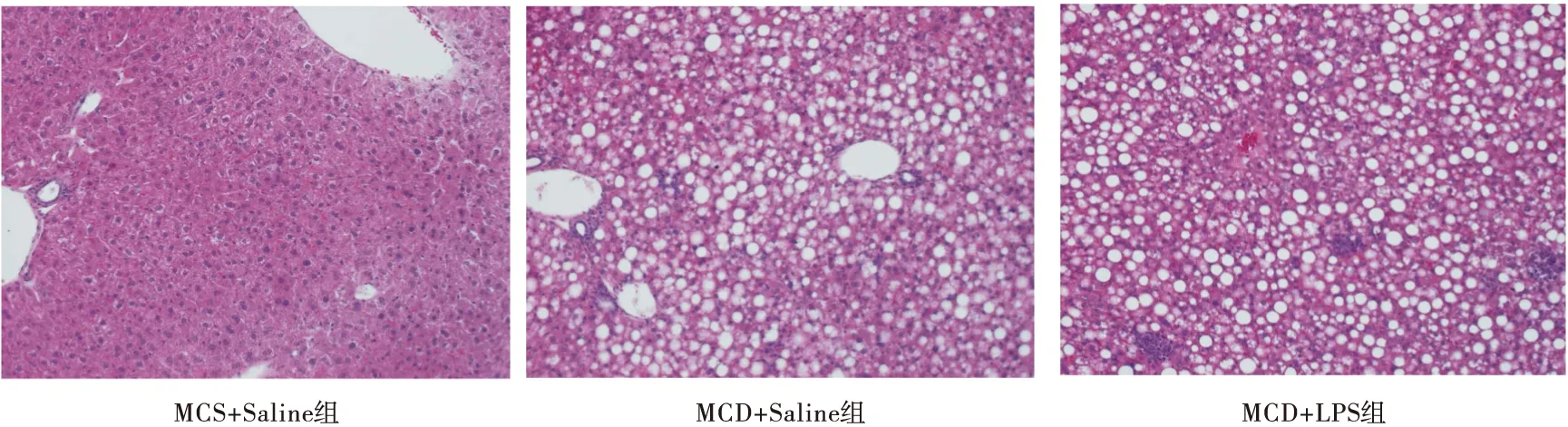
圖1 小鼠肝臟HE染色(×200)
2.2 脂多糖加速NASH小鼠的肝纖維化 通過Sirius Red染色觀察小鼠肝臟的纖維化變化(見圖2)。結果發現,MCS+Saline組除血管外,無明顯紅色著色現象;MCD+Saline組的肝臟組織內可見輕度的纖維化發生;MCD+LPS組的肝臟組織內則可見較多的纖維化發生。

圖2 小鼠肝臟Sirius red染色(×200)
2.3 脂多糖加速NASH小鼠的肝功能受損 血清ALT、AST指標檢測結果見圖3。由圖3可見,與MCS組相比,MCD兩組的血清ALT和AST都顯著升高(P<0.01),另外,腹腔注射LPS進一步使肝功能受損,AST和ALT的含量顯著高于生理鹽水注射組(P<0.05)。
2.4 脂多糖促進NASH小鼠血清中TNF-α的表達 用ELISA試劑盒對血清中TNF-α的含量進行檢測(圖4),結果發現MCD飲食可導致血清TNF-α含量輕度增加(P<0.05),但當注射LPS后,血清中的TNF-α含量增加了約10倍(P<0.01)。
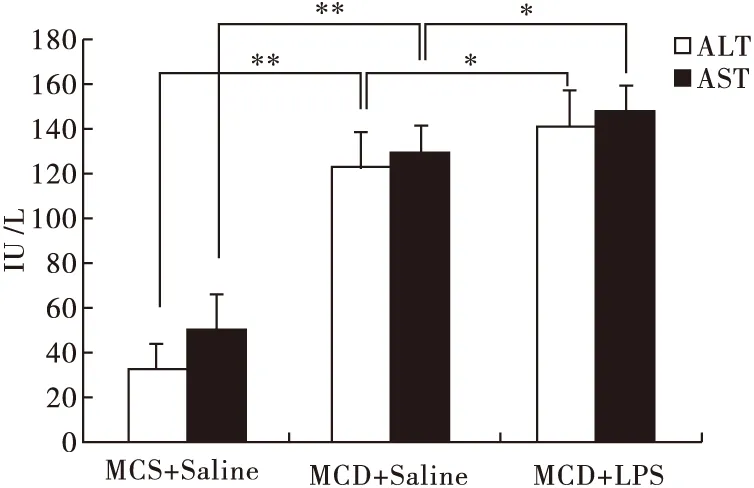
圖3 LPS對NASH小鼠血清ALT和AST的影響

圖4 LPS對NASH小鼠血清TNF-α的影響
2.5 脂多糖進一步加重NASH小鼠肝臟的氧化應激 對肝臟中的TBARS含量進行檢測發現(圖5):與MCS正常飲食組相比,MCD飲食組可造成小鼠肝臟氧化應激水平顯著升高(P<0.01),腹腔注射LPS后進一步加重了肝臟的氧化應激水平(P<0.01)。
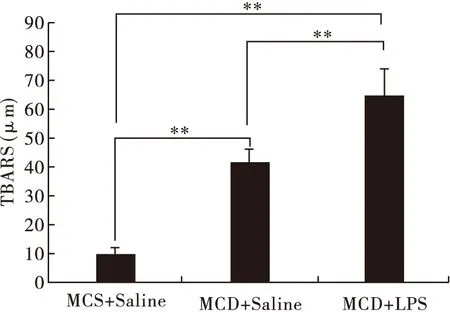
圖5 LPS對NASH小鼠肝臟TBARS的影響
3 討論
近年來研究發現,腸道菌群的移位和紊亂與患者發生慢性肝臟疾病和肝硬化有重要關系[10]。在幾種類型的慢性肝病中,均發現血液中LPS的含量升高。腸道上皮的完整性受損導致其對細菌的通透性改變,因此,腸道細菌發生移位,進入腹腔,之后細菌代謝產生的脂多糖等有害物質加速了肝臟疾病的發展[11]。
本實驗旨在探討LPS對NASH發病進展的研究,因此,我們選用了MCD飲食誘導的NASH小鼠模型。該模型是國際上公認的用于研究NAFLD中炎癥及纖維化的最好的動物模型之一[12]。NASH是NAFLD向非酒精性脂肪性肝硬化(NAC)進展過程中的重要環節,是隱源性肝硬化的重要原因之一[2]。NASH發病機制不明確,目前國際上公認的為“二次打擊學說”[13-14]。肝臟的脂肪變性作為“第一次打擊”,易化肝細胞對其他作為“二次打擊”的因素的敏感性,其中可作為“二次打擊”的物質有很多,包括藥物、超氧陰離子、細胞因子等,這些因素最終導致肝細胞的凋亡及炎性細胞的產生。另有研究發現,NASH患者的肝細胞凋亡程度與病情呈正相關[15-16]。肝細胞凋亡能夠激活肝星狀細胞,進而使之釋放細胞外基質[17-18],這些基質的沉積是引起纖維化發生的根本原因。
本實驗結果顯示,給予小鼠2周的MCD飲食可見明顯的NAFLD癥狀,肝臟內有大量不規則脂肪滴形成,肝組織結構紊亂并出現炎性細胞浸潤,AST和ALT表達增加;當腹腔注射LPS后,炎癥情況增加,并出現壞死前兆,肝纖維化增加,AST和ALT含量進一步增加,肝功能受損加重,并且血清TNF-α含量增加,肝臟氧化應激加重。LPS能夠作用于Kupffer細胞上的CD14和Toll樣受體-4,從而引起TNF-α的合成增多[19]。TNF-α是一種“二次打擊”的因子,能夠使細胞產生氧化應激,使干細胞內產生大量的活性氧集團(ROS),從而進一步導致細胞凋亡[20]。
綜上所述,本實驗在MCD飲食所致NASH小鼠模型的基礎上腹腔注射LPS,引起NASH的病程進展,血清中的TNF-α增加,肝臟氧化應激增加,纖維化增加引起肝功能進一步受損,病程出現不可逆性進展。本研究具有轉化醫學特色,結合臨床發現部分肝病患者服用抗生素后肝功能指標好轉,提示腸道菌群及其釋放的內毒素是加重肝病進展的重要原因之一,希望通過今后更加深入的研究,特別是信號通路方面的進一步探索,為臨床提供治療依據,為患者提供更好的治療方案。
參考文獻:
[1] Roberts EA.Nonalcoholic steatohepatitis in children[J].Curr Gastroenterol Rep,2003,5(3):253-259.
[2] Powell EE,Cooksley WGE,Hanson R,et al.The natural history of nonalcoholic steatohepatitis:a follow-up study of 42 patients for up to 21 years [J].Hepatology,1990,11(1):74-80.
[3] Iimuro Y,Gallucci RM,Luster MI,et al.Antibodies to tumor necrosis factor alfa attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat[J].Hepatology,1997,26(6):1530-1537.
[4] Adachi Y,Moore LE,Bradford BU,et al.Antibiotics prevent liver injury in rats following long-term exposure to ethanol[J].Gastroenterology,1995,108(1):218-224.
[5] Wigg AJ,Roberts-Thomson IC,Dymock RB,et al.The role of small intestinal bacterial overgrowth,intestinal permeability,endotoxaemia,and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis[J].Gut,2001,48(2):206-211.
[6] Li Z,Yang S,Lin H,et al.Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease[J].Hepatology,2003,37(2):343-350.
[7] Loguercio C,De Simone T,Federico A,et al.Gut-liver axis:a new point of attack to treat chronic liver damage[J].Am J Gastroenterol,2002,97(8):2144-2146.
[8] Lee CS,Yan JS,Ng RK,et a1.Polyunsanturated fat in the methinine-choline deficient diet influences hepatic inflammation but not hepatocellular injury [J].J Lipid Res,2007,48 (8):1885-1896.
[9] Fizwm E,Loewno A,Bianca G,et a1.Mice fed a lipogenic methionine choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1[J].J Lipid Res,2006,47(10):2280-2290.
[10]Almeida J,Galhenage S,Yu J,et al.Gut flora and bacterial translocation in chronic liver disease[J].World J Gastroenterol,2006,12(10):1493-1502.
[11] Ilan Y.Leaky gut and the liver:a role for bacterial translocation in nonalcoholic steatohepatitis[J].World J Gastroenterol,2012,18(21):2609-2618.
[12] Su GL.Lipopolysaccharides in liver injury:molecular mechanisms of Kupffer cell activation[J].Am J Physiol Gastrointest Liver Physiol,2002,283(2):256-265.
[13] Basaranoglu M,Basaranoglu G,Sentürk H.From fatty liver to fibrosis:a tale of “second hit”[J].World J Gastroenterol,2013,19(8):1158-1165.
[14]陳興梅.非酒精性脂肪肝患者肝損傷與代謝異常及C反應蛋白的關系[J].中國醫藥,2012,7(4):504.
[15]Wieckowska A,Zein NN,Yerian LM,et a1.In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease[J].Hepatology,2006,44(1):27-33.
[16] Feldstein AE,Canbay A,Angulo P,et a1.Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis[J].Gastroenterology,2003,125(2):437-443.
[17] Kahraman A,Barreyro FJ,Bronk SF,et al.TRAIL mediates liver injury by the innate immune system in the bile duct-ligated mouse[J].Hepatology,2008,47(4):1317-1330.
[18] Canbay A,Friedman S,Gores GJ.Apoptosis:the nexus of liver injury and fibrosis[J].Hepatology,2004,39(2):273-278.
[19]Anstee QM,Goldin RD.Mouse models in non-alcoholic fatty liver disease and steatohepatitis research[J].Int J Exp Pathol,2006,87(1):1-16.
[20] Wei Y,Rector RS,Thyfault JP,et al.Nonalcoholic fatty liver disease and mitochondrial dysfunction[J].World J Gastroenterol,2008,14(2):193-199.
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
中老年保健(2021年3期)2021-08-22 06:50:04
天津醫科大學學報(2021年2期)2021-03-29 05:31:08
現代臨床醫學(2021年1期)2021-01-26 00:56:02
學苑創造·A版(2020年9期)2020-10-13 09:41:02
世界科學技術-中醫藥現代化(2020年2期)2020-07-25 02:05:56
西南軍醫(2016年6期)2016-01-23 02:21:19
西南軍醫(2015年2期)2015-01-22 09:09:37
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
現代檢驗醫學雜志(2014年4期)2014-02-02 02:44:59
