炎癥因子與肝癌干細胞標志在肝癌誘導過程中的相關性研究
2014-08-30 08:59:44宦宏波吳黎靂
實用臨床醫藥雜志 2014年7期
吳 林, 宦宏波, 吳黎靂, 別 平,夏 鋒
(第三軍醫大學西南醫院 全軍肝膽外科研究所, 重慶, 400038)
Kupffer細胞是人體內數量最多的巨噬細胞,約占巨噬細胞總數的80%~90%[1]。它構成了肝臟先天免疫系統的重要組成部分,具有吞噬和殺滅細菌、凋亡細胞、微生物、內毒素、免疫復合物等功能[2-3]; 另一方面, Kupffer細胞在發揮免疫功能的同時釋放白細胞介素6(IL-6)、腫瘤壞死因子α(TNF-α)、γ干擾素(IFN-γ)等炎癥因子參與反應[3-6], 而先前研究證實炎癥反應在腫瘤發生發展過程中起決定性因素[7-9],而且炎癥因子進一步參與了腫瘤干細胞的自我更新和上皮細胞間質轉化(EMT), 促進腫瘤的發生[10-11]。近年來有研究[12-13]報道Kupffer細胞通過釋放炎癥因子參與肝癌的發生發展,但關于Kupffer細胞分泌的炎癥因子和肝癌干細胞標志在肝癌發生發展中的動態表達及其相關性尚未見報道。本研究通過建立大鼠肝癌誘導模型,采用定量PCR方法檢測肝癌發展過程中不同時間點Kupffer細胞分泌的炎癥因子及肝癌干細胞標志的動態變化,并對2者相關性進行統計分析。
1 材料與方法
1.1 材料與試劑
SD雄性大鼠(第三軍醫大學動物中心)、二乙基亞硝胺(DEN, 0.95 g/mL, 美國Sigma公司)、ED2抗體(英國Serotec AbD公司)、磷酸鹽緩沖液(PBS,中杉金橋)、免疫組織化學檢測試劑盒(中杉金橋)、DAB顯色試劑盒(中杉金橋)、Trizol(日本Takara公司)、PrimeScript RT reagent Kit反轉錄試劑盒(日本Takara公司)、SYBR Green(日本Takara公司)、引物合成(北京華大基因公司)。
1.2 儀器與設備
Olympus顯微鏡(日本Olympus公司)、ND1000微量分光光度計(美國Nano Drop公司)、CFX96實時定量PCR儀(美國Bio-Rad公司)。
1.3 試驗方法
1.3.1 肝癌誘導大鼠模型建立: SD雄性大鼠35只(180 g~200 g, 第三軍醫大學動物中心),隨機分為正常對照組(5只),誘導模型組按誘癌時間點分為: 4周、8周、12周、16周、20周、24周(每組5只)。24周組每只大鼠肝臟組織又分為癌和癌旁2個亞組: 24-周-NT(癌旁)、24-周-T(癌)。誘導模型組大鼠按每天10 mL/100 mg body weight飲水中給予DEN溶液(0.01% v/v), 每周僅給予6天DEN溶液,剩余1 d給予普通飲水。正常對照組給予普通飲水。在誘癌過程中,每隔4周取材誘癌模型組5只大鼠的肝臟, -80 ℃冷藏備用,直至誘癌第24周肝癌形成。
1.3.2 免疫組織化學檢測誘癌過程中Kupffer細胞ED2表達:大鼠肝臟組織常規甲醛固定、石蠟包埋、連續切片、烤片后,按免疫組化染色試劑盒說明書進行。脫蠟后, 3% H2O2封閉,抗原修復,山羊血清封閉,一抗ED2(1∶200)4 ℃冰箱過夜,滴加酶標抗體,通用二抗結合, DAB顯色。
各試驗組ED2陽性細胞分數計算方法:每只大鼠計數平均5個高倍鏡視野下每100個細胞中ED2陽性細胞表達分數,取均值和標準差后計算每一組大鼠ED2陽性細胞分數。
1.3.3 實時定量PCR檢測炎癥因子和肝癌干細胞標志表達:所有操作均嚴格按照試劑說明書進行。常規Trizol提取各組大鼠肝臟組織RNA, 測定RNA濃度后,反轉錄試劑盒將mRNA反轉為cDNA,加入特定引物(引物序列見表1),用SYBR Green試劑盒實時定量PCR檢測Kupffer細胞釋放的相關炎癥因子IL-1α、IL-1β、IL-6、IL-8、IL-10、TNF-α、IFN-γ、單核細胞趨化蛋白-1(MCP-1)、轉化生長因子β(TGF-β)和肝癌干細胞指標CD90、CD133、Sirt1、AFP各基因表達, GAPDH作為內參。
1.3 統計學分析
本試驗所有數據均采用SPSS 17.0進行統計分析。誘癌過程中,各試驗組Kupffer細胞ED2陽性分數、Kupffer細胞相關炎癥因子表達和肝癌干細胞標志表達均采用單因素方差分析(ANOVA)和Tukey多重比較。炎癥因子與肝癌干細胞指標之間的相關性采用Pearson相關性檢驗進行分析。P<0.05為差異有統計學意義。
2 結 果
2.1 DEN誘導大鼠肝癌模型的建立
通過對誘導模型組大鼠飲水中加入DEN溶液,大鼠肝癌演變過程表現為經典的炎癥反應期(1~12周)、肝增生纖維化期(12~16周)、肝硬化形成期(16~20周)、肝癌形成期(20~24周)(見圖1, A-N)。到誘癌24周時, 24周組大鼠均發現肝癌形成。

表1 引物合成序列
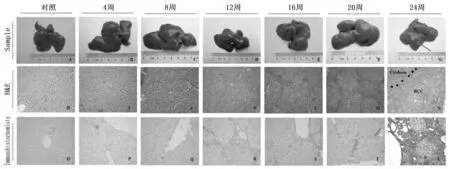
圖1 大鼠肝癌誘導模型建立及Kupffer細胞在誘癌過程中的動態變化100倍
2.2 大鼠肝癌誘導過程中Kupffer細胞ED2表達
通過免疫組化染色檢測Kupffer細胞特異性抗體ED2,觀察大鼠肝癌誘導過程中ED2動態表達,免疫組化結果提示ED2在肝癌誘導過程中表達逐漸增多(圖1, O-U), 并通過統計分析計算出各組ED2陽性細胞分數,結果表明ED2在肝癌誘導過程中呈逐漸升高的趨勢(P<0.001)。而且從誘癌第12周開始,12周、16周、20周和24周的ED2的表達與正常對照組有明顯差異性(P<0.01)。該結果表明Kupffer細胞數量在誘癌過程中呈逐漸增多的趨勢。(圖2)。
2.3 大鼠肝癌誘導過程中Kupffer細胞相關炎癥因子的動態表達
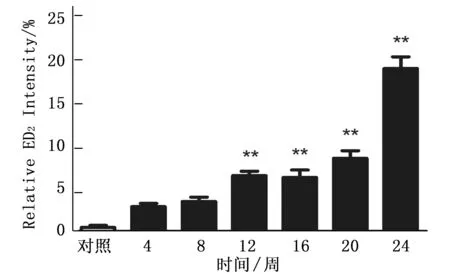
注:**P<0.05
通過定量PCR方法檢測鼠肝癌誘導過程中Kupffer細胞相關炎癥因子的動態表達,結果表明IL-6(P<0.001)、MCP-1(P<0.001)、TGF-β(P=0.003)和TNF-α(P=0.047)在誘癌過程中明顯升高(圖3 A,B,C,D), 而且與正常肝臟組織相比較,在肝癌組織中IL-6(P<0.001)、MCP-1(P<0.001)和TGF-β(P<0.001)表達更高,而其它炎癥因子在誘癌過程中無明顯變化(圖3, E-I,P>0.05)。
2.4 大鼠肝癌誘導過程中肝癌干細胞標志的動態表達

注:**P<0.05
通過定量PCR方法檢測鼠肝癌誘導過程中肝癌干細胞標志的動態表達,結果表明CD90在誘癌過程中呈逐漸升高的趨勢(P<0.001),特別是與正常肝臟組織比較,肝癌組織中CD90升高更明顯(圖4 A;P<0.001)。CD133雖然呈一定升高趨勢,但ANOVA分析其結果無明顯變化(P=0.125), 同時其它肝癌干細胞標志物在誘癌過程中無明顯變化(圖4 B,C,D;P>0.05)
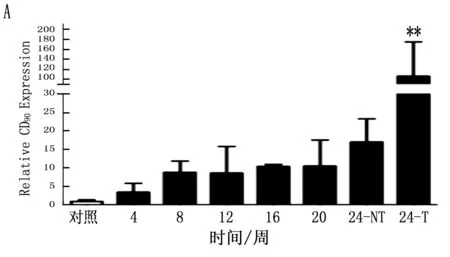
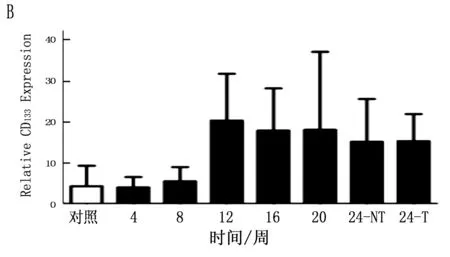
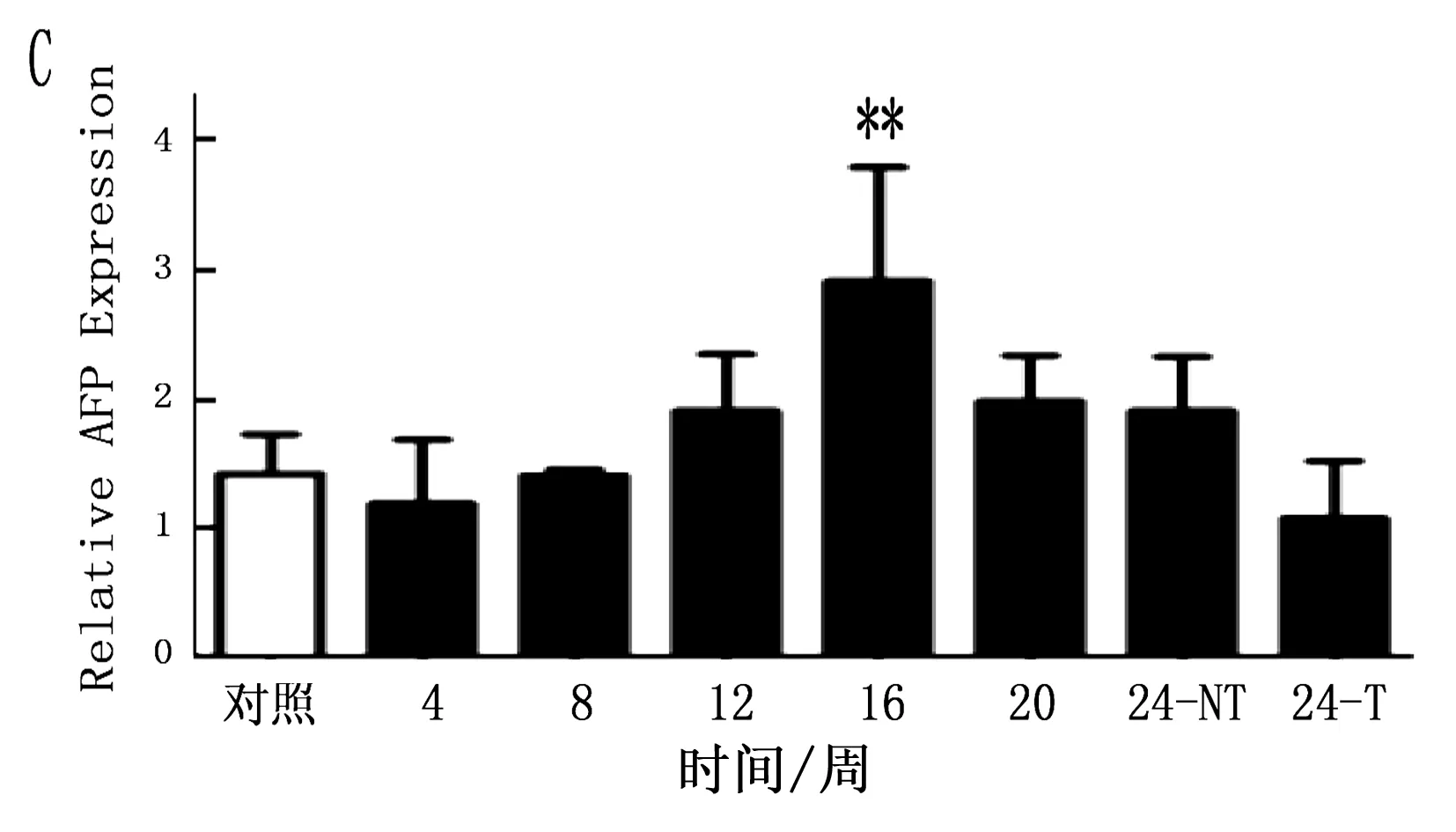
注:**P<0.05
2.5 Kupffer相關炎癥因子與肝癌干細胞標志的相關性
通過以上結果統計分析篩選出在誘癌過程中差異性表達的Kupffer相關炎癥因子: IL-6、MCP-1、TGF-β和TNF-α, 以及差異性表達肝癌干細胞標志CD90。將肝癌誘導過程中CD90與IL-6、MCP-1、TGF-β和TNF-α的動態表達進行Pearson相關性分析,結果表明IL-6、MCP-1、TGF-β和CD90的表達呈明顯正相關性(圖5 A,B,C;P<0.001), 而TNF-α與CD90表達無明顯相關性(圖5 D;P=0.288)。
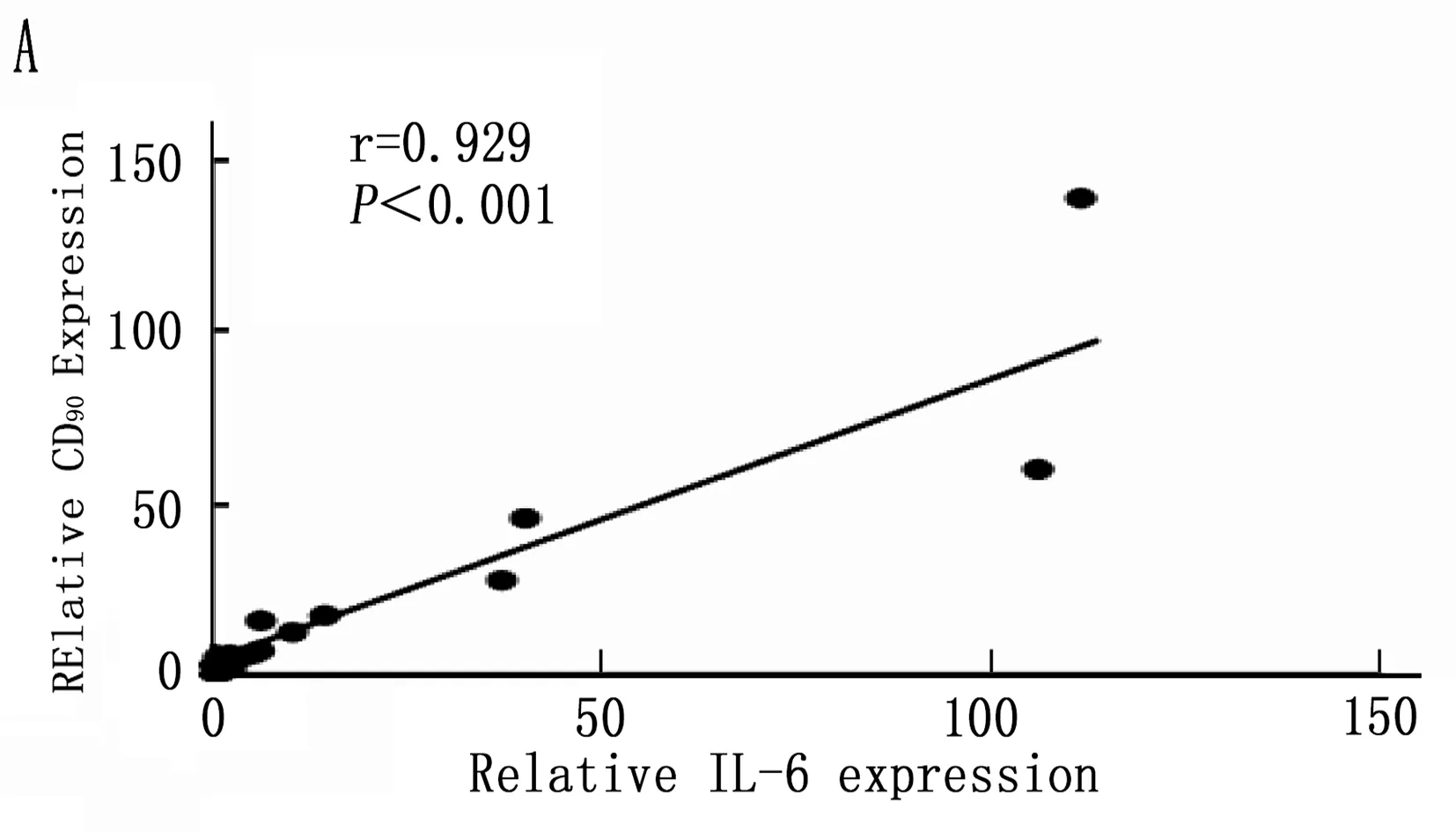
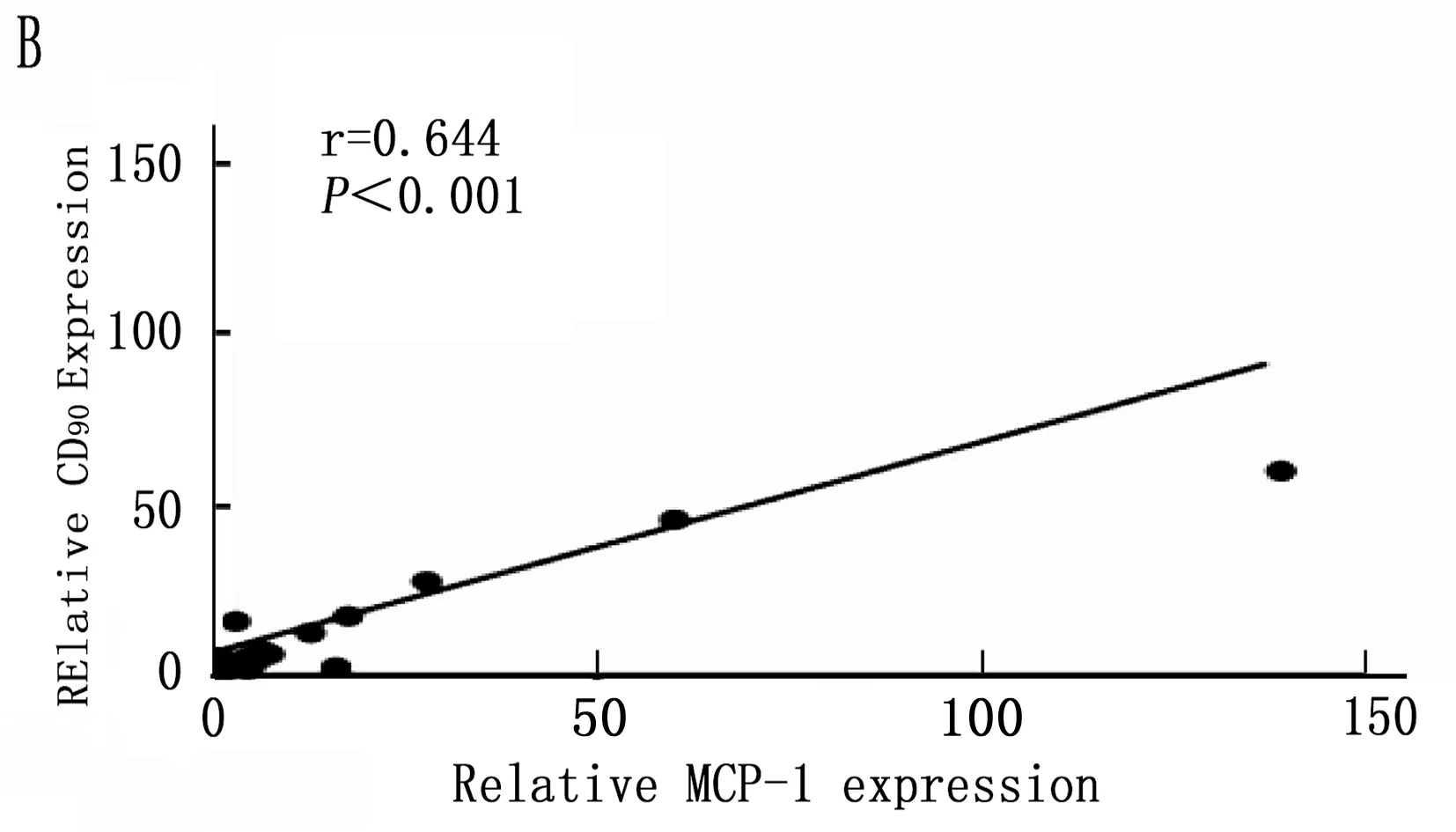
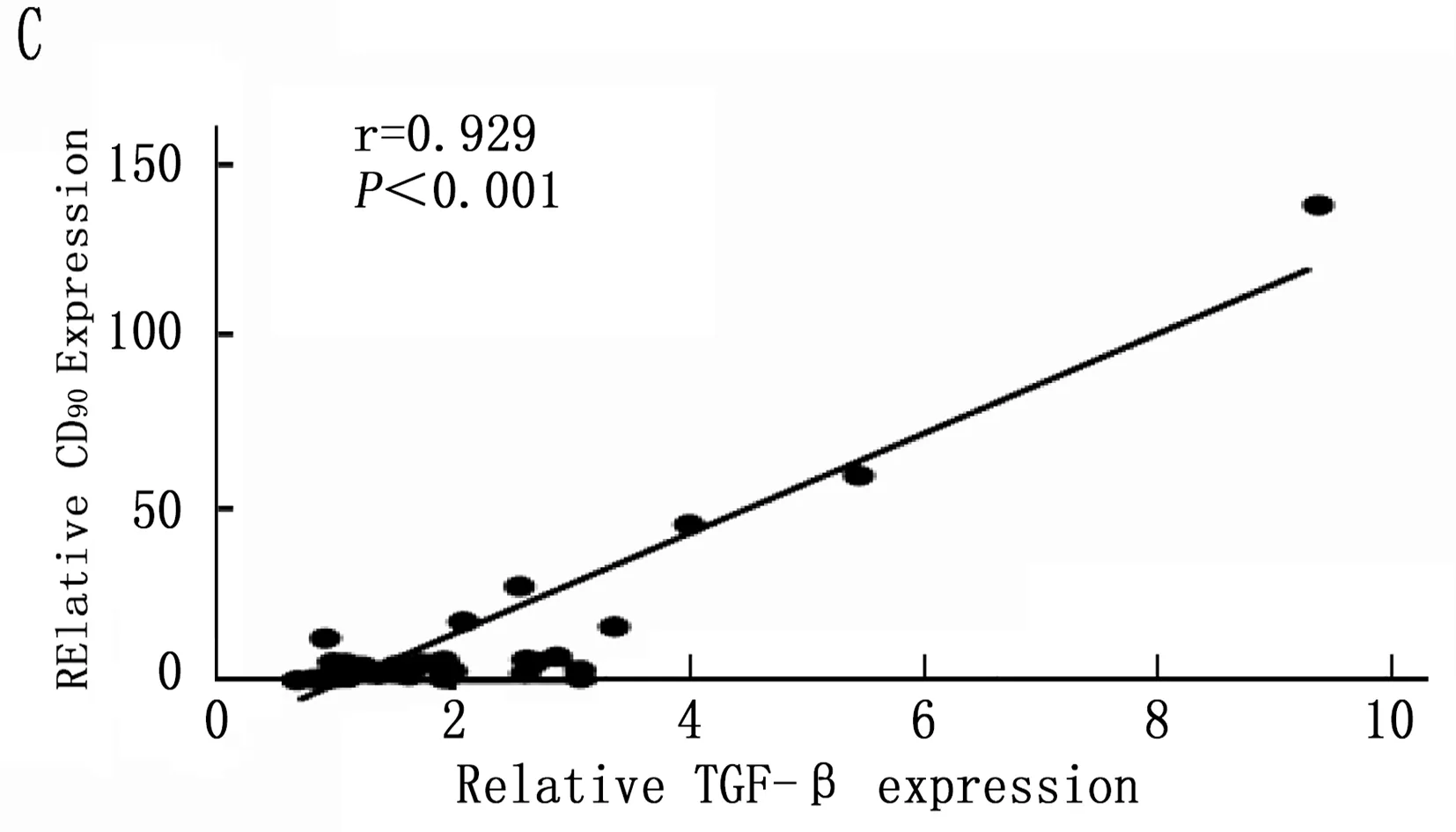
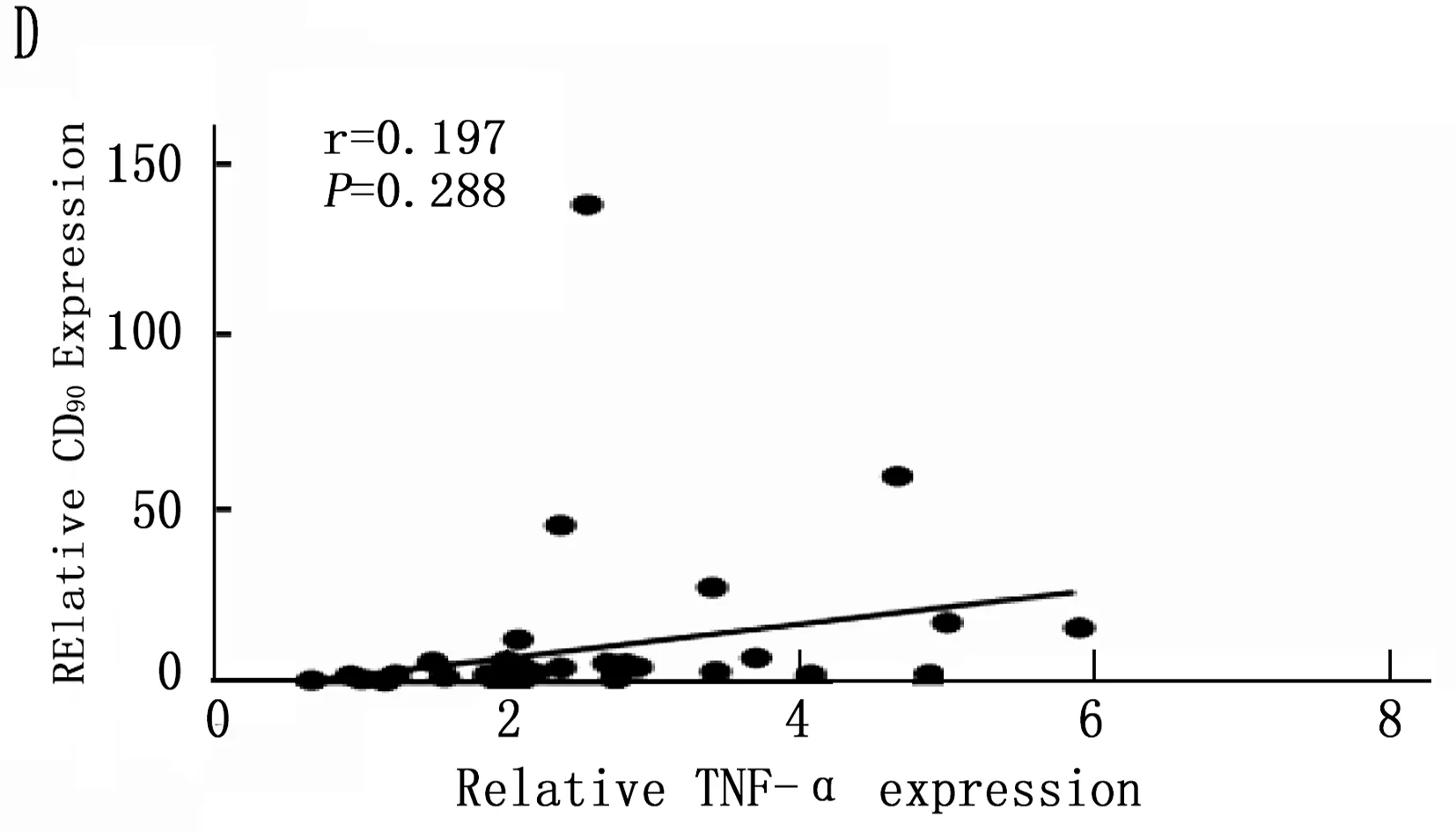
圖5 肝癌誘導過程中CD90與Kupffer細胞相關炎癥因子的相關性分析
3 討 論
本研究首先通過DEN誘導成功建立經典大鼠肝硬化肝癌模型,在誘癌24周時發現肝臟腫瘤,然后對各組大鼠采用免疫組織化學方法檢測了Kupffer細胞的特異性抗體ED2[14-15]在肝癌誘導過程中的動態變化,結果表明ED2陽性細胞在誘癌過程中逐漸升高(圖2;P<0.001)。采用定量PCR方法檢測了Kupffer細胞釋放的炎癥因子在誘癌過程中的表達變化,結果篩查出IL-6(P<0.001)、MCP-1(P<0.001)、TGF-β(P=0.003)和TNF-α(P=0.047)在誘癌過程中變化明顯(圖3 A,B,C,D); 同時肝癌干細胞標志物篩查出CD90指標在誘癌過程中呈明顯升高趨勢(圖4 A;P<0.001)。通過Pearson檢驗分析IL-6、MCP-1、TGF-β、TNF-α與CD90的相關性,提示IL-6、MCP-1、TGF-β與CD90的表達明顯相關(圖5 A,B,C;P<0.001)。從而證明Kupffer細胞產生的炎癥因子與肝癌的發生發展密切相關。
許多研究證實炎癥因子與腫瘤發生之間存在密切關系。Naugler WE等[12]通過研究DEN對不同性別大鼠肝癌形成的影響,證明雌性大鼠通過雌激素調控Kupffer細胞上Toll樣受體銜接蛋白MyD88,抑制壞死肝細胞釋放IL-6以及清除循環血液中的IL-6, 從而達到抑制化學性誘癌的發生。Wu K等[16]在一部分大鼠及人體肝癌組織中發現肝臟前體細胞共表達腫瘤初始細胞標志(T-IC), 同時TGF-β的表達與T-IC的表達呈正相關性,而在缺乏TGF-β的情況下卻不能啟動肝癌的發生,從而證明了TGF-β在肝癌發生發展中的重要作用。Wang WW等[17]通過蛋白抗體芯片檢測了58例肝癌切除患者及11例乙肝病毒攜帶者的血清蛋白,結果發現113種血清標志物被調控,其中MCP-1的變化最為明顯,而且MCP-1有希望作為AFP的一種補償標志物,通過ELISA常規定量檢測患者血清MCP-1濃度,從而幫助臨床預測和診斷肝癌。Yang ZF等[18]對肝癌患者循環血液及肝癌組織進行研究發現, CD45-、CD90+細胞可以在90%的肝癌患者循環血液及肝癌組織中被檢測,證明CD90可以作為檢測和診斷肝癌的一個重要標志物。
部分研究也明確了炎癥因子與腫瘤干細胞之間的相關性。Jin X等[10]證明在腦腫瘤發生過程中,干擾素調節因子7(IRF-7)可以通過IL-6和Notch信號通路誘導神經膠質瘤細胞的產生以及血管生成。此外, Li Y[11]等通過研究結腸癌細胞,發現IL-1β可以通過激活腫瘤干細胞的自我更新及EMT促進結腸癌的生長與侵襲。Korkaya H等[19]對曲妥珠單抗的耐藥乳腺癌細胞系研究發現, IL-6炎癥反饋通路導致了腫瘤干細胞的擴增,長時間通過曲妥珠單抗處理的細胞系篩選出大量的腫瘤干細胞,這些腫瘤干細胞表現出EMT現象及其分泌的IL-6相比普通細胞系表達高出近百倍,同時通過阻斷IL-6受體可以明顯降低腫瘤的生長和轉移。以上試驗均表明炎癥因子對腫瘤干細胞的重要調控作用,而Kupffer細胞釋放的相關炎癥因子與肝癌干細胞標志CD90的相關性尚未見報道。
本研究尚存在一些不足之處: ① Kupffer細胞相關炎癥因子促進肝癌干細胞標志表達的具體機制還需進一步功能學試驗明確; ②本試驗結果篩查出CD90與Kupffer細胞釋放的炎癥因子明顯相關,但其它肝癌干細胞標志物與炎癥因子的相關性還需進一步探討。因此,作者設想下一步是否可以通過抑制Kupffer細胞釋放的炎癥因子,或將Kupffer細胞清除達到降低甚至抑制肝癌的發生。
綜上所述,本研究結果表明Kupffer細胞釋放的炎癥因子IL-6、MCP-1、TGF-β的表達升高與CD90上調呈明顯正相關,進一步明確了Kupffer細胞促進肝癌發生的重要作用。因此, Kupffer細胞可以作為潛在的肝癌治療靶點為肝癌的臨床診治開辟新的領域。
[1]Liaskou E, Wilson D V, Oo YH. Innate immune cells in liver inflammation[J]. Mediators Inflamm, 2012, 2012: 949157.
[2]Kolios G, Valatas V, Kouroumalis E. Role of kupffer cells in the pathogenesis of liver disease[J]. World J Gastroenterol, 2006, 12(46): 7413.
[3]Laskin D L. Macrophages and Inflammatory Mediators in Chemical Toxicity: A Battle of Forces[J]. Chem Res Toxicol, 2009, 22(8): 1376.
[4]Helk E, Bernin H, Ernst T, et al. TNFα-mediated liver destruction by Kupffer cells and Ly6Chi monocytes during Entamoeba histolytica infection[J]. PLoS Pathog, 2013, 9(1): e1003096.
[5]Lucey, M. R., P. Mathurin, et al. Alcoholic hepatitis[J]. N Engl J Med, 2009, 360(26): 2758.
[6]Xu L, Yin W, Sun R, et al. Kupffer cell-derived IL-10 plays a key role in maintaining humoral immune tolerance in hepatitis B virus-persistent mice[J]. Hepatology, 2013, doi: 10.1002/hep.26668.[Epub ahead of print]
[7]Grivennikov S I, Greten F R, Karin M. Immunity, Inflammation, and Cancer[J]. Cell, 2010, 140(6): 883.
[8]Park E J, Lee J H, Yu G Y, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression[J]. Cell, 2010, 140(2): 197.
[9]Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker[J]. Nat Immunol, 2011, 12(8): 715.
[10]Jin X, Kim S H, Jeon H M, et al. Interferon regulatory factor 7 regulates glioma stem cells via interleukin-6 and Notch signalling[J]. Brain, 2012, 135(Pt 4): 1055.
[11]Li Y, Wang L, Pappan L, et al. IL-1β promotes stemness and invasiveness of colon cancer cells through Zeb1 activation[J]. Mol Cancer. 2012, 11: 87.
[12]Naugler W E, Sakurai T, Kim S, et al. Gender Disparity in Liver Cancer Due to Sex Differences in MyD88-Dependent IL-6 Production[J]. Science, 2007, 317(5834): 121.
[13]Maeda S, Kamata H, Luo J L, et al. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis[J]. Cell, 2005, 121(7): 977.
[14]Tapia G, Santibá?ez C, Farías J, et al. Kupffer-cell activity is essential for thyroid hormone rat liver preconditioning[J]. Mol Cell Endocrinol, 2010, 323(2): 292.
[15]Golbar H M, Izawa T, Murai F, et al. Immunohistochemical analyses of the kinetics and distribution of macrophages, hepatic stellate cells and bile duct epithelia in the developing rat liver[J]. Exp Toxicol Pathol, 2012, 64(1/2): 1.
[16]Wu K, Ding J, Chen C, et al. Hepatic Transforming Growth Factor Beta Gives Rise to Tumor-Initiating Cells and Promotes Liver Cancer Development[J]. Hepatology, 2012, 56(6): 2255.
[17]Wang W W, Ang S F, Kumar R, et al. Identification of serum monocyte chemoattractant protein-1 and prolactin as potential tumor markers in hepatocellular carcinoma[J]. PLOS ONE, 2013, 8(7): e68904.
[18]Yang Z F, Ngai P, Ho D W, et al. Identification of Local and Circulating Cancer Stem Cells in Human Liver Cancer[J]. Hepatology, 2008, 47(3): 919.
[19]Korkaya H, Kim G I, Davis A, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population[J]. Mol Cell. 2012, 47(4): 570.
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
天津醫科大學學報(2019年3期)2019-08-13 06:53:08
中成藥(2016年8期)2016-05-17 06:08:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
腫瘤預防與治療(2015年1期)2015-09-26 07:26:20
中國當代醫藥(2015年16期)2015-03-01 02:03:11
