鎮咳寧顆粒的質量標準研究
2014-09-11 02:18:57
中國民族民間醫藥 2014年8期
貴陽濟仁堂藥業有限公司,貴州 貴陽 550005
鎮咳寧顆粒的質量標準研究
李興勇
貴陽濟仁堂藥業有限公司,貴州 貴陽 550005
目的建立鎮咳寧顆粒的質量標準。方法采用薄層色譜法鑒別鎮咳寧顆粒中甘草、鹽酸麻黃堿;采用高效液相色譜法對制劑中麻酸麻黃堿進行定量分析,色譜條件為:Hypersil C18(250×4.6 mm,5 μm)柱,甲醇-(0.1%磷酸溶液︰0.1三乙胺溶液(1︰1))9︰91為流動相,流速為1.0 ml/min,柱溫40 ℃,檢測波長210 nm,進樣量為10 μL。結果薄層色譜法能對甘草、鹽酸麻黃堿進行專屬性定性分析,鹽酸麻黃堿進樣量在77.0~616.0 ng(r=0.9999)范圍內與峰面積積分值線性關系良好;平均加樣回收率為100.75%,RSD為0.46%(n=6)。結論所建立的方法能準確地進行定性和定量檢測,方法快速簡便、準確可靠、重復性好,可有效的控制鎮咳寧顆粒的質量。
鎮咳寧顆粒;甘草;鹽酸麻黃堿;TLC;HPLC
鎮咳寧顆粒是由衛生部藥品標準《中成藥成方制劑》第三冊收載的“鎮咳寧糖漿”(WS3-B-0665-91)[1]改劑型而成,由甘草流浸膏、桔梗、鹽酸麻黃堿、桑白皮組成,具有鎮咳祛痰的功效。臨床用于傷風咳嗽,支氣管炎,哮喘等。而哮喘、咳嗽多于夜間發作,其原因主要由于支氣管平滑遇冷或者受到刺激而引起支氣管收縮痙攣誘發哮喘、咳嗽。針對這一原因,筆者對方中藥味進行了研究。麻黃堿為擬腎上腺素藥,能松弛支氣管平滑肌、收縮血管,有顯著的中樞興奮作用,臨床主要用于治療習慣性支氣管哮喘和預防哮喘的發作,有很好的療效。但鹽酸麻黃堿過量,對高血壓、心率失常的患者有較大的危害,需嚴格控制其用量,故對其進行定量測定的研究控制具有重要意義。本研究在原“鎮咳寧糖漿”的基礎上,經過多次摸索,增訂了方中甘草的薄層色譜鑒別,同時采用高效液相色譜法對方中鹽酸麻黃堿進行了定量分析研究,獲得了滿意效果,為建立完整的質量標準提供了快速、簡便、準確的定性定量方法。
1 儀器與試藥
1.1 儀器 LC-15C高效液相色譜儀(日本島津公司);WML-2010威瑪龍色譜數據工作站(南寧市威瑪龍色譜科技有限公司);UV-1750型紫外可見分光光度計(日本島津公司);AUW120D型電子天平(日本島津公司);KQ-500VDB雙頻數控超聲波清洗機(昆山市超聲儀器有限公司);電熱恒溫水浴鍋(天津市泰斯特儀器有限公司)。
1.2 試藥 甘草對照藥材(批號:120904-200914),鹽酸麻黃堿對照品(批號:171214-201007,供含量測定用以99.7%計,使用前需經105 ℃干燥3 h),均由中國食品藥品檢定研究院提供;鎮咳寧顆粒(批號:20121101,20121102,20121103,20121104,20121201,20121202,20121203,20121204,20121205,20121206),由貴陽濟仁堂藥業有限公司研制;甲醇(美國迪馬公司生產);乙腈(美國迪馬公司生產)為色譜純,水為二次重蒸餾水;其余試劑均為分析純。
2 薄層色譜鑒別
2.1 鹽酸麻黃堿的薄層色譜鑒別 取本品6g,加水10ml和氨試液5ml使溶解,溶液用乙醚-三氯甲烷-乙醇(25∶8∶2.5)混合液30ml振搖提取,靜置,分取上層液,加酸性乙醇(取乙醇20ml,加鹽酸1ml,混勻)1ml,蒸干,殘渣加甲醇1ml使溶解,作為供試品溶液。另取鹽酸麻黃堿對照品,加甲醇制成每1ml含1mg的溶液,作為對照品溶液。照薄層色譜法(中國藥典2010年版一部附錄Ⅵ B)試驗,吸取上述兩種溶液各5μl,分別點于同一以羧甲維素鈉為黏合劑的硅膠G薄層板上,以三氯甲烷-甲醇-濃氨試液(20∶3.5∶0.5)為展開劑,展開,取出,晾干,噴以印三酮試液,在105℃加熱約5 min。供試品色譜中,在與對照品色譜相應的位置上,顯相同的紫紅色斑點。見圖1。

1 2 3 4 5
2.2 甘草的薄層色譜鑒別 取本品8g,加甲醇30ml浸漬2h,并時時振搖,濾過,濾液揮干,殘渣加甲醇1ml使溶解,作為供試品溶液。另取甘草對照藥材2g,加稀乙醇30ml浸漬4h,濾過,濾液揮干,殘渣加甲醇1ml使溶解,作為對照藥材溶液。照薄層色譜法(中國藥典2010年版一部附錄Ⅵ B)試驗,吸取上述兩種溶液各5~10μl,分別點于同一以羧甲基纖維素鈉為黏合劑的硅膠G薄層板上,以三氯甲烷-甲醇-水(13∶7∶2)的下層液為展開劑,展開,取出,晾干,噴10%硫酸乙醇溶液,105 ℃加熱至斑點清晰。供試品色譜中,在與對照藥材色譜相應的位置上,顯相同顏色斑點。見圖2。
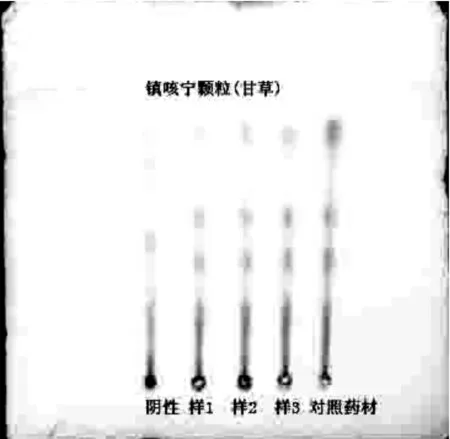
1 2 3 4 5
3 鹽酸麻黃堿的含量測定
3.1 色譜條件 色譜柱:Hypersil C18(250×4.6 mm,5 μm)柱;柱溫:40 ℃;流動相:甲醇-(0.1%磷酸溶液︰0.1三乙胺溶液(1︰1))(9︰91);流速:1.0 ml/min;檢測波長:210 nm;理論板數:按鹽酸麻黃堿峰計應不低于3000。
3.2 溶液的制備
3.2.1 對照品溶液的制備 取鹽酸麻黃堿對照品15.40 mg,置100 ml量瓶中,加稀乙醇溶解并稀釋至刻度,搖勻,即得鹽酸麻黃堿對照品貯備液(每1 ml含0.1540 mg)精密吸取20ml對照品貯備液,置100ml容量瓶中,加稀乙醇稀釋至刻度,搖勻,即得對照品溶液(每1 ml含30.80 μg鹽酸麻黃堿)。
3.2.2 供試品溶液的制備 取裝量差異項下的本品內容物適量,混勻,研細,取約0.5g,精密稱定,加入稀乙醇25 ml,稱定重量,回流提取30 min,取出,放冷,再稱定重量,用稀乙醇補足減失的重量,搖勻,用微孔濾膜(0.45μm)濾過,取續濾液,即得。
3.2.3 陰性對照溶液的制備 取處方中除鹽酸麻黃堿外的其他藥材,按0.1倍處方劑量及制法和工藝要求制備缺鹽酸麻黃堿的陰性樣品,照供試品溶液的制備方法制成缺鹽酸麻黃堿陰性對照溶液。
3.3 方法專屬性考察 分別吸取對照品溶液、供試品溶液和陰性對照溶液各10μL,注入HPLC儀,測定,見色譜圖3。由圖3可見,陰性對照在與對照品相同保留時間(約為9.5 min)的位置上,無相應的色譜峰出現,表明除鹽酸麻黃堿外的其他藥材和輔料對測定無干擾,且鹽酸麻黃堿與相鄰雜質峰能夠很好的分度,分度度大于1.5。
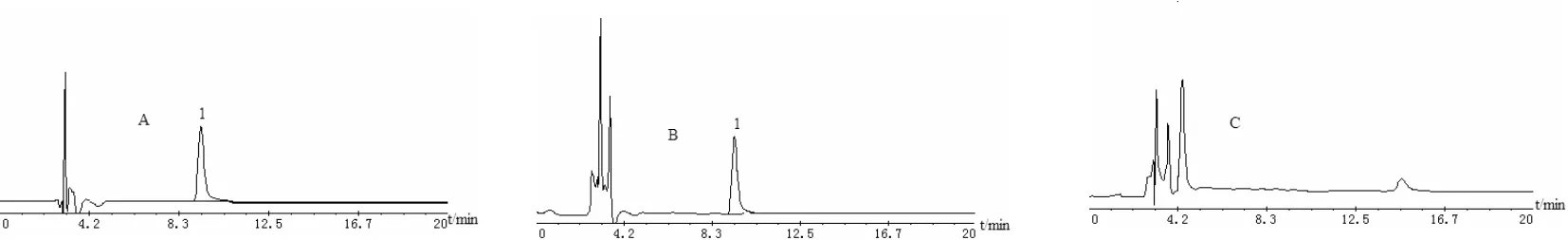
圖3 鎮咳寧顆粒HPLC圖
3.4 線性關系的考察 精密吸鹽酸麻黃堿對照品貯備液(0.1540 mg/ml)7份,分別制成7.70,15.40,23.10,30.80,38.50,46.20,61.60μg/ml的溶液,精密吸取上述溶液各10μl,注入液相色譜儀,按上述“3.1”項下的色譜條件進行試驗,以峰面積為縱坐標,進樣量(μg)為橫坐標繪制標準曲線,鹽酸麻黃堿回歸方程為Y = 1 240.859 X-52.156(r=0.999 9),擬合為過原點的回歸方程為Y=1 240.732 X(r=0.9999)。結果表明,鹽酸麻黃堿在進樣量為77.0~616.0ng范圍內具有良好的線性關系。
3.5 精密度試驗 精密吸取鹽酸麻黃堿對照品溶液(濃度為30.80μg/ml)適量,每次10μl,重復進樣測定 6 次,記錄峰面積,平均峰面積為380 355,峰面積RSD為0.26%,表明具有良好的精密度。
3.6 穩定性試驗 取樣品適量,按“3.2.2”項下方法制備供試品溶液,室溫下分別在0,1,2,4,6,8,10h各取10μl注入高效液相色譜儀,記錄峰面積,結果峰面積平均值為376 144,峰面積的RSD為0.47%,表明在室溫下10h內供試品溶液具有良好的穩定性。
3.7 重復性試驗 取樣品適量,混勻,研細,精密稱取樣品6份,按“3.2.2” 項下方法制備供試品溶液進行測定,各取10μl注入高效液相色譜儀,記錄峰面積,計算鹽酸麻黃堿的含量,結果樣品中鹽酸麻黃堿的平均含量為1.815 mg/g,RSD為0.52%,說明有良好的重復性。
3.8 加樣回收率試驗 取樣品(含量為1.815 mg/g)適量,混勻,研細,取6份,每份約0.25 g,精密稱定,置具塞錐形瓶中,精密加入鹽酸麻黃堿對照品溶液15ml(濃度為30.80μg/ml),稀乙醇10ml,按“3.2.2”項下方法制備供試品溶液,進樣測定峰面積,計算加樣回收率,結果見表1。
3.9 樣品含量測定 按“3.2.2”項下方法制備供試品溶液,精密吸取對照品溶液和供試品溶液各10μL,注入液相色譜儀,測定峰面積,計算樣品中鹽酸麻黃堿的含量,10批樣品中鹽酸麻黃堿測定結果見表2。
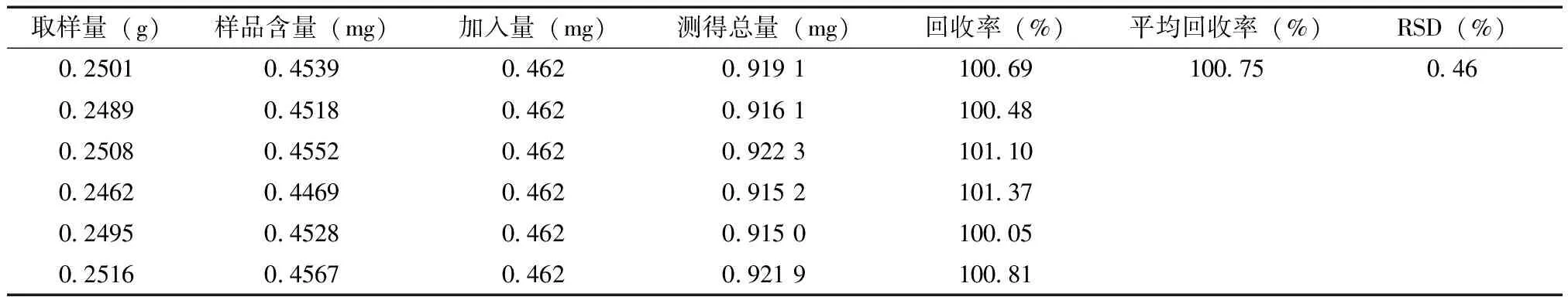
表1 鎮咳寧顆粒中鹽酸麻黃堿加樣回收率
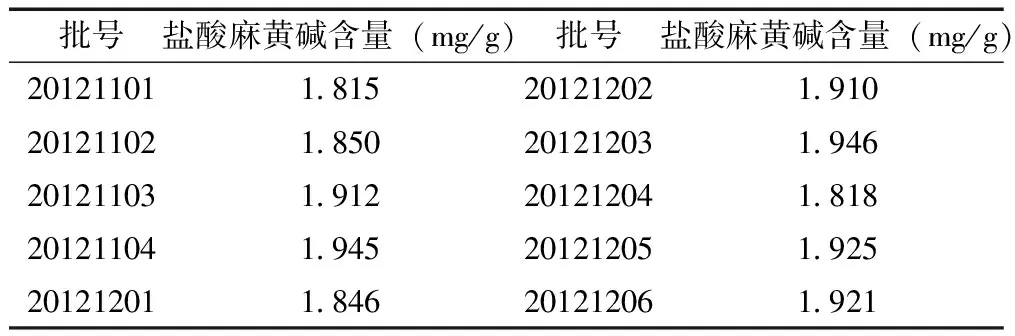
表2 鎮咳寧顆粒中鹽酸麻黃堿含量
4 討論
參照文獻[2],筆者對樣品提取方法進行了研究,分別以甲醇、50%甲醇、0.01%鹽酸甲醇、乙醇、稀乙醇為提取溶劑進行試驗。結果表明,以稀乙醇作為溶劑時峰形好、柱效高、回收率好,故選用稀乙醇作為提取溶劑。
鎮咳寧顆粒薄層色譜鑒別研究表明,本研究的TLC方法可用于鎮咳寧顆粒的TLC鑒別;HPLC方法學考察結果表明,本法操作較為簡單,準確性、重復性好,可作為鎮咳寧顆粒的含量測定方法。
從10批樣品的含量測定結果可以看,鹽酸麻黃堿的平均含量均在標示量的90.0%~110.0%之間,表明本法測定具有較好的準確性。
[1]衛生部藥典委員會.中華人民共和國衛生部藥品標準中成藥成方制劑(第3冊).WS3-B-0665-91[S].北京:化學工業出版社,1998:203.
[2]國家藥典委員會.中華人民共和國藥典.一部[S].北京:中國醫藥科技出版社,2010:300,485,489,495,496,504.
李興勇(1974-),男,學士,研究方向:中藥質量標準的研究。
R284.1
A
1007-8517(2014)08-0021-03
2014.01.24)
