新型固體酸催化芐基甲苯的合成
2014-12-31 12:30:16孔令杰張東恒李鵬李洪偉官婷婷
潤滑油 2014年1期
關鍵詞:催化劑
孔令杰,張東恒,李鵬,李洪偉,官婷婷
(中國石油大連潤滑油研究開發中心,遼寧大連 116032)
0 引言
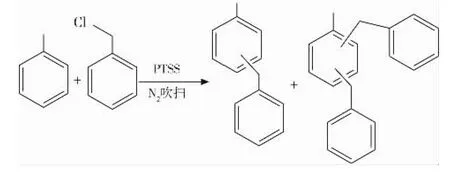
芐基甲苯是一類重要的化工產品。其中二芐基甲苯因具有熱穩定性好、黏度低、導熱系數高和安全性好等優點被廣泛用于高溫導熱油的調制[1-2]。此外,單芐基甲苯和二芐基甲苯特定比例的混合物還是一種優良的電力電容器浸漬劑,廣泛應用于電力行業[3-4]。
芐基甲苯的合成主要是通過甲苯和氯化芐的傅克烷基化反應,一般需要加入催化劑對反應進行催化。所采用的催化劑一般有兩類,一類是路易斯酸催化劑,如 AlCl3、FeCl3、BF3、TiCl4等;另一類是液態質子酸催化劑,如 H2SO4、H3PO4等。由于質子酸類催化劑對反應釜的腐蝕較大,所以傳統工業上采用較多的是AlCl3等均相催化劑[5-11],但此類催化劑也存在難以和反應產物分離等問題。目前研究報道比較多的是采用固體超強酸對此反應進行催化[12-13]。固體超強酸具有催化活性好、不腐蝕設備和易與產物分離等優點。但所見報道中鮮有對此類反應具有很高活性的固體超強酸催化劑,氯化芐的轉化率不能令人十分滿意。
本文采用自主開發的固體超強酸催化劑PTSS對甲苯和氯化芐的傅克烷基化反應進行了研究。探討了反應溫度、反應配比、反應時間及催化劑用量等因素對反應的影響,并對分離得到的二芐基甲苯進行了理化性能和熱穩定性測試。
1 實驗
1.1 催化劑的制備、表征
催化劑前體為金屬水合氧化物,經過硫酸浸泡、水洗、過濾和焙燒等環節制備而成。
NH3-TPD表征采用ChemBet TPD自動升溫化學吸附儀,實驗程序為:稱取200 mg試樣置于石英U型管中,用石英棉封好并與系統相連。He氣流下以10℃/min的速率升溫至400℃并恒溫60min,然后降溫至100℃,在此溫度下NH3吸附60 min,然后He氣吹掃60 min,最后以10℃/min的速率升溫至800℃進行程序升溫脫附實驗。
1.2 反應的過程與方法
在一定的溫度、攪拌和氮氣吹掃下,向已加入甲苯和催化劑PTSS的反應體系中緩慢滴加一定摩爾比的氯化芐。反應產生的氯化氫在氮氣的攜帶下通入酸吸收裝置。反應結束后通過減壓蒸餾獲取目標產物。
1.3 產物組成的測定與表征
反應粗產物采用Agilent7890氣相色譜儀進行分析,分析條件:進樣口溫度350℃,檢測器溫度350℃,色譜柱型號HP-5,升溫程序從110~320℃,升溫速率20℃/min。
產物的結構通過H1NMR進行表征,核磁共振儀為Varian INOVA 400MHz。
2 實驗結果與討論
2.1 PTSS的NH3-TPD表征
圖1是PTSS的NH3-TPD譜圖。
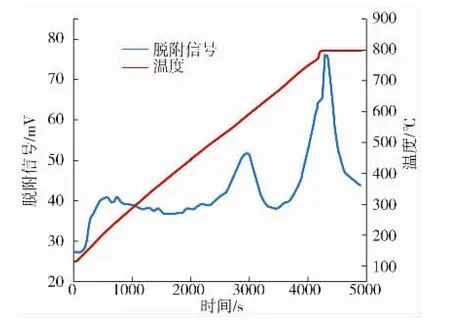
圖1 催化劑PTSS的NH3-TPD譜圖
NH3-TPD數據顯示(見圖1),催化劑PTSS為典型的超強酸型固體催化劑,200℃左右的脫附峰顯示其弱酸中心的存在,550℃左右的脫附峰顯示其中強酸中心的存在。在800℃左右時,催化劑PTSS釋放強的脫附信號,表明其超強酸中心的存在。
2.2 反應物配比對反應的影響
固定反應的溫度為100℃,反應時間為5 h,催化劑加劑量為氯化芐的0.5%,考察了反應物配比對反應的影響,實驗結果見圖2。數據顯示,制備的PTSS超強酸催化劑對甲苯與氯化芐的烷基化反應具有明顯的催化效果。氯化芐的轉化率隨甲苯與氯化芐摩爾比的增大呈現先增大后減小的趨勢,當甲苯與氯化芐摩爾比為3∶1時,轉化率最大,達到95.4%,當甲苯與氯化芐摩爾比增大為5∶1時,氯化芐的轉化率僅為76%。這與催化劑被稀釋有一定關系。很多文獻給出優化的甲苯與氯化芐的摩爾比為5∶1以上,并且隨摩爾比的增大,氯化芐轉化率也增大。顯然,這種變化趨勢的差異是由所使用的催化劑導致的。文章采用的催化劑能夠在盡量小的甲苯與氯化芐摩爾比的條件下得到高的氯化芐的轉化率,縮小了反應體系總量,為反應的工業化提供了便利。同時,反應后處理階段需要蒸出的甲苯量降低,能夠節約大量的能耗成本。

圖2 甲苯與氯化芐的摩爾比對氯化芐轉化率的影響
通過GC的分析可知,當甲苯與氯化芐的摩爾比為3∶1時,反應產物中的單芐基甲苯與二芐基甲苯的摩爾比為5∶1,產物中沒有多芐基甲苯的生成。而當甲苯與氯化芐的摩爾比降低為2∶1時,雖然反應產物中單芐基甲苯與二芐基甲苯的摩爾比為2.7∶1,二芐基甲苯的摩爾比例提高,且氯化芐的轉化率也達到94.6%,但此時已有少量的多芐基甲苯出現,因此將優化的投料比定為3∶1。
2.3 反應溫度對反應的影響
固定甲苯與氯化芐的摩爾比為3∶1,反應時間為5 h,催化劑加劑量為氯化芐的0.5%,考察了反應時間對反應的影響,實驗結果見圖3。氯化芐的轉化率受溫度影響較大,當反應溫度為80℃時,氯化芐的轉化率僅為22%。當反應溫度提升至100℃以上時,氯化芐的轉化率明顯提升,達到90%以上。同時,二芐基甲苯在產物中的比例也隨之由80℃時的不到1%提升至120℃時的21%。反應溫度提升至120℃時,氯化芐的轉化率最大,但此時有少量的多芐基甲苯的生成。因此將反應的優化溫度定為甲苯的回流溫度110℃,此時氯化芐的轉化率為96.2%。

圖3 反應溫度對氯化芐轉化率的影響
2.4 反應時間對反應的影響
固定甲苯與氯化芐的摩爾比為3∶1,反應溫度為110℃,催化劑加劑量為氯化芐的0.5%,考察了反應時間對反應的影響,實驗結果見圖4。

圖4 反應時間對氯化芐轉化率的影響
隨著反應時間的增長,氯化芐的轉化率逐漸增大。在反應的初始階段(1~3 h),反應進行迅速,氯化芐轉化率增速明顯。反應進行3 h之后,隨著反應體系中氯化芐的濃度降低,氯化芐轉化率增速變緩。當反應時間增長至5 h以后,反應進行十分緩慢,因此,確定優化的反應時間為5 h。
2.5 催化劑用量對反應的影響
固定甲苯與氯化芐的摩爾比為3∶1,反應溫度為110℃,反應時間為5 h,考察了催化劑用量對反應的影響,實驗結果見圖5。氯化芐的轉化率隨催化劑用量的增加而增加,當催化劑用量達到氯化芐質量的0.8%后,氯化芐的轉化率達到99%。繼續增大催化劑用量,轉化率基本不變。催化劑用量越大,單位體積內其催化位點越多,則氯化芐的轉化率越大。繼續增大催化劑的量雖然有利于產物中二芐基甲苯比例的增大,但另一方面會導致多芐基甲苯的生成。因此,確定優化的催化劑用量為氯化芐質量的0.8%。

圖5 催化劑用量對氯化芐轉化率的影響
2.6 產物的結構與表征
通過減壓蒸餾獲取目標化合物單芐基甲苯和二芐基甲苯,并進行了H1NMR表征。
圖6為單芐基甲苯的H1NMR,可以看出主要有三種類型的氫,δ=2.2-2.3(d,CH3,3H),δ=3.93-3.97(d,CH2,2H),δ =7.07-2.28(m,ArH,9H),這三類的氫的峰面積之比為 3∶2∶9,氫的位移以及比例均與單芐基甲苯一致。從圖譜上看,甲基氫和亞甲基氫是雙重峰,事實上是兩種不同的異構體導致的。由傅克烷基化反應的電子效應規律可知,單芐基甲苯應該主要有鄰、對位取代產物兩種。因此,推測得到的兩種單芐基甲苯為鄰、對位取代產物。

圖6 單芐基甲苯的H 1 NMR
圖7為二芐基甲苯的H1NMR,可以看出也主要有三種類型的氫,δ=2.08-2.29(m,CH3,3H),δ=3.93-3.97(m,CH2,4H),δ =7.07-2.28(m,ArH,13H),這三類的氫的峰面積之比為3∶4∶13,氫的位移以及比例均與二芐基甲苯一致。由于二芐基甲苯的前體是單芐基甲苯,而單芐基甲苯已存在異構體,所以生成的二芐基甲苯存在多種異構體,導致氫譜的化學位移出現細微差別。

圖7 二芐基甲苯的H1 NMR
2.7 二芐基甲苯的理化性能及其作為導熱油的指標
將分離純化后的二芐基甲苯進行了理化性能和熱穩定性能的測試,結果顯示,其各項性能能夠很好滿足國標GB 23971-2009中關于有機熱載體的各項指標要求,試驗數據見表1。
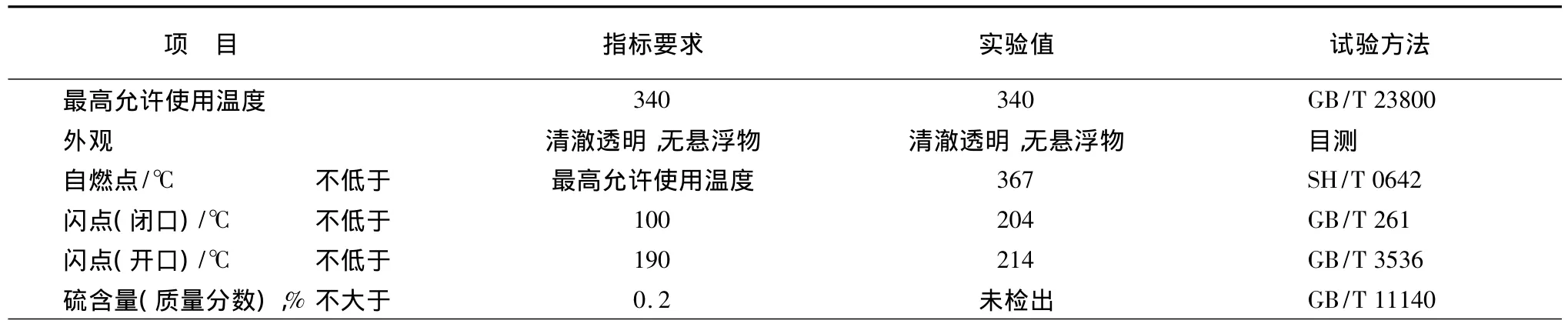
表1 二芐基甲苯的技術要求和實測值

續表
3 結論
自主開發的固體超強酸催化劑PTSS對甲苯與氯化芐的傅克烷基化反應具有良好的催化活性,能夠在相對小的反應體系中獲取優異的反應結果。優化的反應條件為:反應溫度110℃、反應時間5 h、甲苯與氯化芐的摩爾比為3∶1、催化劑用量為氯化芐用量的0.8%。在優化條件下,氯化芐的轉化率可達到99%以上。合成的二芐基甲苯各項理化指標和熱穩定性能均能很好地滿足國標GB 23971-2009有機熱載體的指標要求,具有良好的應用前景。
[1]陳聲宗,陳欣,陳俊彥,等.用二芐基甲苯代替氫化三聯苯作高溫導熱油的研究[J].合成纖維,1997(3):13-15.
[2]Winfried G,Essen,Anto Benning.Method of Carrying out Reactions Using Liquid Heat Transfer Agents:US,3475115[P].
[3]Noelle Berger,Raymond Commandeur,Pierre Jay.Dielectric Compositions Comprising Benzyltoluene/(Methylbenzyl)Xylene Isomers:US,5545355 A[P].
[4]Keiji Endo,Shigenobu Kawakami,Atsushi Sato.Electrical Insulating Oil Composition and Capacitors Prepared Therewith:US,5081758 A[P].
[5]Yusuke I,Mayumi O,Kazuo U,et al.Salts and Ammonium Salts of Keggin-Type Heteropolyacids as Solid Acid Catalysts for Liquid-Phase Friedel-Crafts Reactions[J].Appl Catal A,1995,132:127-140.
[6]Vasant R C,Suman K J.Benzylation of Benzene and Substituted Benzenes by Benzyl Chloride over InCl3,GaCl3,FeCl3and ZnCl2Supported on Clays and Si-MCM-41[J].JMol Catal A:Chem ,2002,180:267-276.
[7]Bachari K,Millet JM M,Benachouba B,etal.Benzylation of Benzene by Benzyl Chloride over Iron MesoporousMolecular Sieves Materials[J].JCatal,2004,221:55-61.
[8]Chaube V D.Benzylation of Benzene to Diphenylmethane U-sing Zeolite Catalysts[J].Catal Commun,2004,5:321-326.
[9]Tivadar C,S ?ndor B,Francois F,et al.Benzylation of Aromatics on Lon-Exchanged Clays[J].JMol Catal A:Chem ,1995,98:101-107.
[10]Salavati-Niasari M,Hasanalian J,Najafian H.Alumina-Supported FeCl3,MnCl2,CoCl2,NiCl2,CuCl2,and ZnCl2as Catalysts for the Benzylation of Benzene by Benzyl Chloride[J].JMol Catal A:Chem,2004,209:209-214.
[11]Vasant R C,Suman K J,Ajit SM.Benzylation of Benzene by Benzyl Chloride over Fe-Modified ZSM-5 and H-B Zeolites and Fe2O3or FeCl3Deposited on Micro-,Meso-and Macro-Porous Supports[J].Microporous Mesoporous Mater,2002,56:65-71.
[12]季山,廖世軍.SO2-4/ZrO2類固體超強酸的研究進展[J].石油化工,2000,29(9):701-703.
[13]王知彩,孫正俊,等.固體超強酸催化劑催化甲苯與氯化芐的芐基化反應[J].石油化工,2005,34(10):954-958.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
