云南中華按蚊的遺傳變異和種群結構
2015-01-18 07:43:08楊飛龍李旭東閆振天付文博
生態學報 2015年16期
楊飛龍,李旭東,閆振天,付文博,陳 斌
重慶師范大學生命科學學院,昆蟲與分子生物學研究所, 重慶 401331
云南中華按蚊的遺傳變異和種群結構
楊飛龍,李旭東,閆振天,付文博,陳 斌*
重慶師范大學生命科學學院,昆蟲與分子生物學研究所, 重慶 401331
為了掌握云南省各地中華按蚊種群間的遺傳變異和種群結構特征,測序并分析了采自云南9個樣本點5個種群組的89頭中華按蚊的線粒體COII基因。結果表明這些中華按蚊種群的COII基因序列平均單倍型多樣性指數和核苷酸多樣性指數分別為h=0.933,π=0.00406,共有51個變異位點,占分析的739個堿基總數的6.9%;定義了39個單倍型,有2個頻率最高的單倍型H1和H9,分別占個體序列數的20.2%和12.4%;系統發育分析表明單倍型與地理位置沒有明顯的對應關系,單倍型網絡圖顯示大部分單倍型分布沒有明顯的親緣地理格局,主要以單倍型H1、H9、H4、H33和H2為中心呈星狀分布,但元江和元陽構成的種群組(YU)單倍型存在明顯地域分布特征;AMOVA結果表明種群組間遺傳變異為12.58%,達到顯著水平(P=0.04888),地理種群組間具有明顯種群遺傳結構。不同地區兩兩種群組間的Fst值和Nm值顯示大部分種群組間存在基因交流,沒有形成明顯的遺傳分化,但YU種群組和其他種群組間缺乏明顯的基因交流,這主要是因為哀牢山的阻隔,使云南東西部形成兩種不同的氣候,產生了明顯的遺傳分化;歧點分布圖顯示為明顯單峰分布,中性檢測結果均為顯著負值,說明云南省的中華按蚊種群在近期經歷過復雜的種群擴張事件。掌握中華按蚊遺傳多樣性及分化特征,對中華按蚊及瘧疾控制具有重要的作用。
云南省;中華按蚊;mtDNACOII;遺傳變異;種群結構
中華按蚊(Anophelessinensis)是按蚊亞屬赫坎按蚊種團中研究比較多的蚊種,它廣泛分布于東亞[1- 3],在我國分布于東經100—120°,北緯19—54°[4],屬于按蚊亞屬中的優勢種。近年來,中華按蚊被認為是瘧疾特別是間日瘧的重要傳播媒介[5],亞洲很多地區都有其瘧疾傳播的報道。在韓國,該種被認為是重要的傳瘧媒介[6- 7],主要傳播間日瘧[8- 9]。在日本,中華按蚊分布比較廣泛,一直被認為是日本溫帶地區(包括沖繩及北海道)最重要的瘧疾傳播媒介[2- 3]。在我國,中華按蚊更多的被認為是間日瘧原蟲的傳播媒介而非惡性瘧原蟲的傳播媒介[4,10],特別是我國的云南省,由于地處熱帶和亞熱帶地區,中華按蚊種群受季節影響較小;同時云南省水稻田分布廣泛,致使中華按蚊成為其分布最廣泛、最主要的傳瘧媒介之一;加之云南和緬甸、老撾、越南接壤,常年境外瘧疾的輸入,使瘧疾防治工作變得十分困難,進一步加大了云南瘧疾的流行趨勢。
盡管中華按蚊具有重要醫學意義,但關于其種群遺傳結構相關的報道卻很少。種群遺傳結構的研究有助于闡明自然條件下物種內的遺傳變異,及其與地理環境的關系以及物種形成的機理,加深對物種進化過程的認識[11],從而對物種的防治或保護提供重要科學依據。目前,蚊蟲的控制主要依賴殺蟲劑的使用,理解種群的遺傳分化有助于推斷殺蟲劑抗性的現狀和可能擴散途徑,從而指導殺蟲劑的使用。馬雅軍對我國部分中華按蚊群體分子遺傳多態性進行了研究[12],云南省相對于中國其他地區,擁有其獨特的氣候條件和復雜的地理環境,中華按蚊種群結構具有一定差別。因此,本研究通過收集云龍、騰沖、盈江、中緬邊境、雙江、景洪、勐臘、元江和元陽9個采集點的中華按蚊標本,分析其線粒體COII序列,進一步對云南的中華按蚊遺傳變異和種群結構進行了研究。
1 材料和方法
1.1 材料
本研究的樣本為中華按蚊野外成蚊,共89頭,采于2010年10月到2012年9月,采自云南省中緬邊境、盈江、景洪、勐臘、雙江、騰沖、元江、元陽和云龍9個采樣點,并根據9個采樣點的地理位置間的距離劃分為5個種群組(表1,圖1)。采集后的樣本用85%酒精保存在-20℃冰箱中。
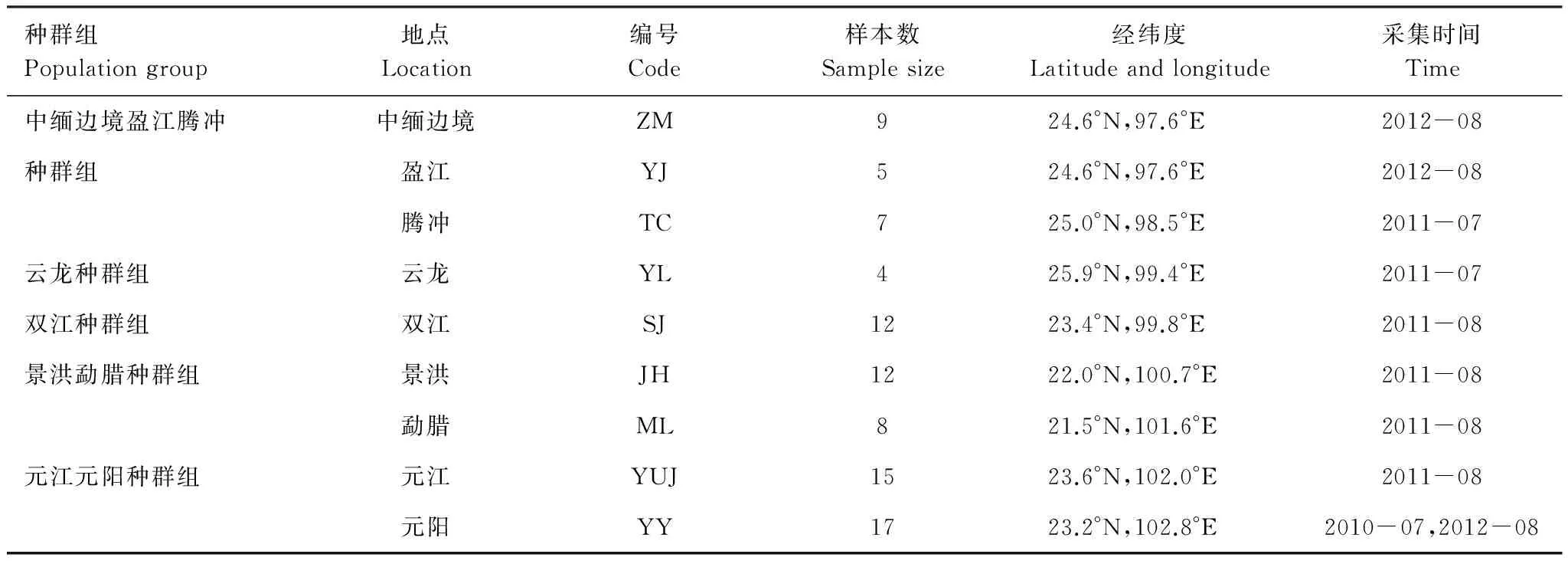
表1 云南省中華按蚊不同種群樣本采集情況
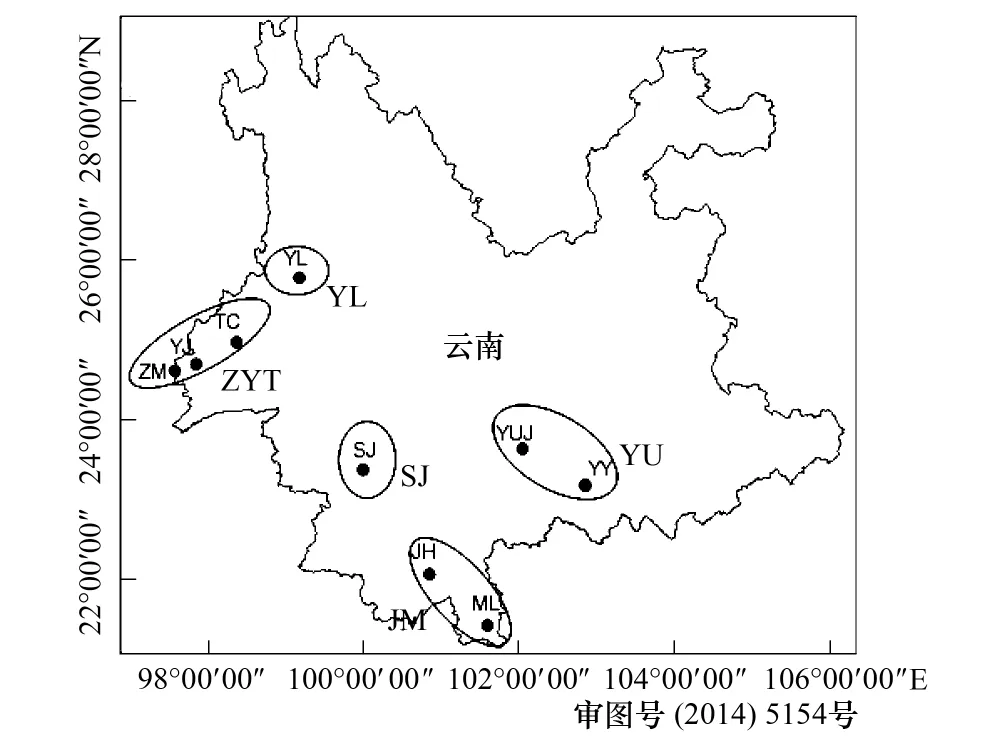
圖1 云南省中華按蚊9個采樣點及5個種群組的地理位置 Fig.1 Nine collecting localities of An. sinensis populations in Yunnan, and five population groups grouped based on their geographical distributionsZYT: 中緬邊境盈江騰沖種群組; YL: 云龍種群組; SJ: 雙江種群組; JM: 景洪勐臘種群組; YU: 元江元陽種群組
1.2 方法
1.2.1 形態學鑒定
形態學上中華按蚊的主要鑒定部位包括觸須、翅脈、腹部和后足跗節。本研究根據董學書編著的《云南蚊蟲志》檢索表[13],在顯微鏡下對中華按蚊標本主要部位進行鑒定,篩選出中華按蚊。
1.2.2 DNA提取
根據QIAGEN試劑盒使用說明提取中華按蚊基因組DNA。提取步奏:(1)組織樣品與 Buffer ATL混合,磨碎,再加入蛋白酶K,56 ℃水浴過夜。(2)再依次加入Buffer AL、無水乙醇,震蕩混勻。(3)將混勻液加入到DNeasy Mini spin column中,6000r/min離心1 min。(4)將spin column轉移到新的收集管中,加入Buffer AW1,6000r/min離心1 min。(5)將spin column轉移到新的收集管中,加入Buffer AW2,20000r/min離心3 min。(6)將柱子轉移到1.5 mL的離心管中。(7)加入Buffer AE到柱子膜上,在室溫下放置1 min,在6000r/min下離心分離1 min。(8)重復第7步,然后將提取的DNA放于-20 ℃冰箱保存備用。
1.2.3 分子鑒定
由于形態學鑒定存在一定誤差,因此將提取的DNA樣品,進行分子鑒定。擴增28S rDNA ITS2片段,擴增引物,D1:5′-TGTGAACTGCAGGACACATGAA- 3′,D2:5′-AGGGTCAAGGCATACAGAAGGC- 3′[14]。PCR反應總體積為25 μL,PCR擴增的反應體系:模板DNA 1 μL,正反引物(10 μmol/L)各1 μL,再加入Taq Mastermix到25 μL。反應程序:95 ℃預變性5 min,95 ℃變性40 s,55 ℃退火40 s,72 ℃延伸1 min,35個循環后,72 ℃延伸6 min。PCR產物進行電泳檢測,結果為目標條帶1077 bp的PCR產物,送上海生物工程有限公司測序,將得到的序列放入NCBI進行比對。
1.2.4 COII基因PCR擴增和測序
對分子鑒定為中華按蚊的樣品,進行COII基因PCR擴增。擴增引物,LEU (forward): 5′-TCTAATATGGCAGATTAGTGCA- 3′ 和LYS (reverse): 5′-ACTTGCTTTCAGTCATCTAATG- 3′[15]。PCR反應總體積為25 μL,PCR擴增的反應體系:模板DNA 1 μL,正反引物(10 μmol/L)各1 μL,再加入Taq Mastermix到25 μL。反應程序:95 ℃預變性5 min,95 ℃變性1 min,51 ℃退火1 min,72 ℃延伸2 min,35個循環后,72 ℃延伸10 min后4 ℃保存。PCR產物用1.5%瓊脂糖凝膠電泳檢測樣品擴增情況,對成功擴增的樣品送上海生工生物工程有限公司進行雙向測序。
1.3 數據分析
使用MEGA5.1[16]軟件將測序得到的序列進行同源比對和構建單倍型鄰接(NJ)樹,Bootstrap重復抽樣1000次檢驗。用DnaSP 5.0[17]軟件計算各地理中華按蚊種群的單倍型多樣型指數(Haplotype diversity,h)、核苷酸多樣性指數(Nucleotide diversity,π)、多態性位點個數(Polymorphic sites,s)[18],繪制單倍型歧點分布圖[19],計算種群間的遺傳分化Fst值和基因交流Nm值。利用Arlequin v3.1[20]中的分子變異分析(AMOVA),根據pairwise difference模型,進行分子方差分析,評估種群內與種群間的遺傳分化,并進行中性檢測(Tajima′sD檢測和Fu & Li檢測),以檢測云南省中華按蚊種群是否符合中性變異。應用Network 4.0[21]構建單倍型網絡結構圖,根據不同地理關系分析各種群之間的進化關系,以追溯單倍型間的親緣關系。
2 結果與分析
2.1 中華按蚊形態和分子鑒定
從形態學上初步篩選出中華按蚊標本95頭,對95頭中華按蚊標本開展分子鑒定,有89頭標本電泳結果產生了明顯的中華按蚊條帶(圖2)。將擴增的89份PCR產物測序后使用Blast在NCBI數據庫中比對,序列與NCBI數據庫中的已知中華按蚊28S rDNA ITS2序列相似度為99%—100%。

圖2 中華按蚊分子鑒定PCR產品電泳圖 Fig. 2 The agarose gel electrophoresis of PCR product for molecular identification of An. sinensis
2.2 COII測序及序列變異與多樣性
對獲得的89條中華按蚊COII序列進行拼接比對,在這些序列中共檢測到39個單倍型,占總樣本數89的43.8%。A、T、C、G的平均含量分別為:35.99%、39.51%、12.58%、11.91%,A+T含量(75.37%)明顯高于G+C含量(24.63%),COII堿基組成具有很大的偏向性,G的含量很低。在所分析的序列中,共檢測到變異位點51個(圖3),占分析位點總數739 bp的6.9%,其中簡約信息位點17個,單一突變位點34個。在不同的群體中變異位點最多的是元江元陽種群組,有24個,變異位點最低的是云龍種群組,有7個。
總體的單倍型多樣性指數較高,但核苷酸多樣性指數較低(表2)。其中,云龍種群組單倍型多樣性指數最高為1.000,采集到4頭標本,分屬4個單倍型,但核苷酸多樣性不高,說明個體間遺傳差異較小;其次是景洪勐臘種群組單倍型指數為0.937,中緬邊境盈江騰沖種群組最低,為0.890。云龍種群組核苷酸多樣性也是最高,為0.00496,說明相對于其他種群個體間遺傳差異較大;其次是景洪勐臘種群組,為0.00429;元江元陽種群組最低,為0.00406。所有單采集樣點種群作為一個種群進行分析,單倍型多樣性指數為0.931,核苷酸多樣性指數為0.00466。
89頭中華按蚊標本COII基因共檢測到39個單倍型。其中H1、H2、H9分布最廣,除了云龍種群組外,這些單倍型在其余種群組均有分布,可能是在云龍種群組采樣數目較少,單倍型類型相應少。H1單倍型頻率最高,為20.2%(18/89),而H2、H9單倍型頻率分別為6.7%和12.4%;H12主要分布在元江元陽種群組和少部分的景洪勐臘組種群,頻率為7.9%;H3分布于中緬邊境盈江騰沖種群組和景洪勐臘種群組,頻率為4.5%;其他單倍型頻率都很低,其中有29個單倍型為種群獨有單倍型,分布于所研究的5個種群組,各個地區獨有單倍型的多少可能跟采集樣本的多少或者跟本身地理環境引起的遺傳變異有關。
2.3 系統發育分析和單倍型網絡關系
用MEGA構建的NJ樹(圖4)顯示單倍型系統發育關系的支持率大部分很低,且主要分為兩個大枝,分別由12個單倍型和27個單倍型組成,但每個大枝都涉及5個種群組的單倍型分布,單倍型拓撲結構并沒有顯示出與地理位置相對應關系的聚集,這說明云南的中華按蚊沒有形成明顯的地理遺傳變化。

圖3 中華按蚊COII基因變異位點Fig.3 Variation sites of the COII gene in An. sinensis
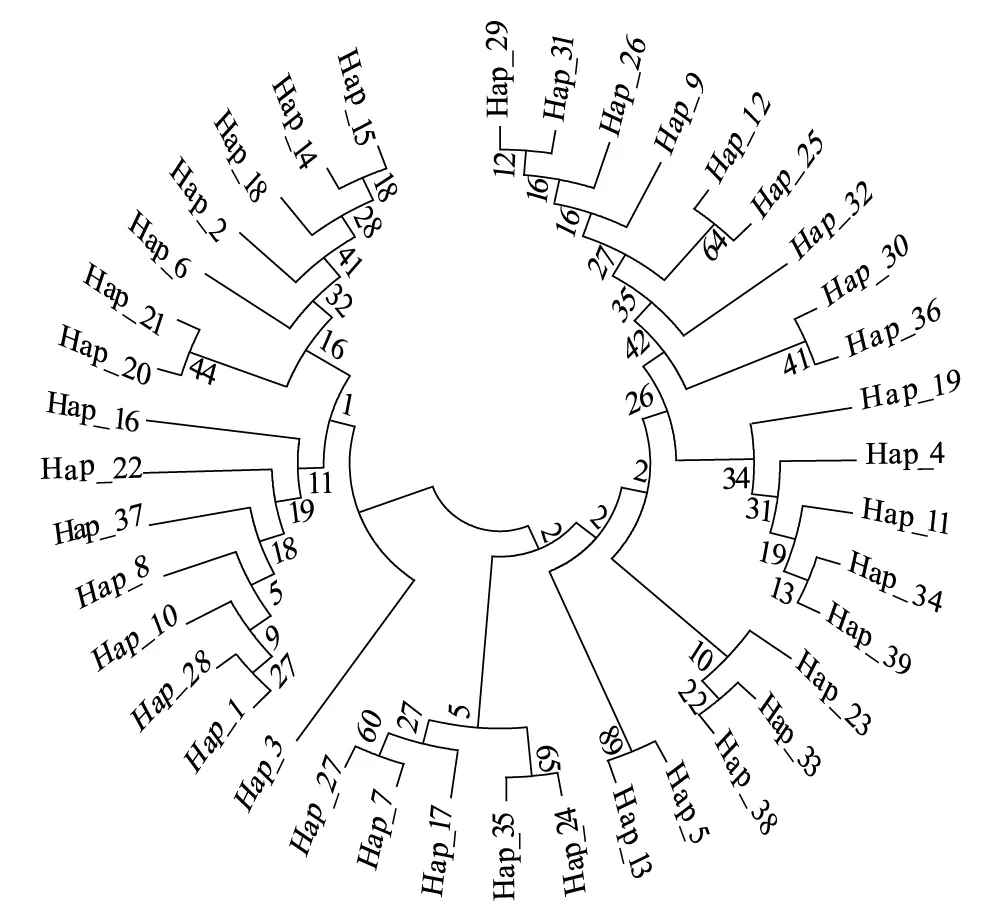
圖4 中華按蚊COII序列NJ樹Fig.4 NJ tree of An. sinensis COII sequences

表2 中華按蚊樣本采集地、樣本數、單倍型及變異統計
用Network的Median-joining方法構建的單倍型網絡圖(圖5),圖中餅圖大小與單倍型出現頻率成正比。由圖可知大部分種群地理位置與單倍型間沒有顯示出明顯的對應關系,但元江元陽種群組單倍型存在與地理位置明顯的對應關系。所有單倍型以星狀網絡結構進行分布,H1、H9、H4、H33和H2是主要的單倍型,其他單倍型以這5個單倍型為中心,輻射分布開來。其中H1單倍型占據主導優勢,可能是這些單倍型中最為原始的單倍型,其他單倍型由其進化而來。
2.4 種群間遺傳分化和種群結構分析
將云南的9個采集地點劃分為5個中華按蚊種群組進行分子變異分析,結果顯示種群組間遺傳變異為12.58%,達到顯著水平(P=0.04888)(表3),說明地理種群組間具有明顯種群遺傳結構。Fst值顯示(表4)大部分種群間沒有出現明顯的遺傳分化現象,但元江元陽種群組除了和云龍種群組沒有遺傳分化外和其余3個種群都存在明顯的遺傳分化,同時Nm值顯示元江元陽種群組除了和云龍種群組存在明顯的基因交流外和其他種群的基因交流并不明顯,兩組數據反映結果一致,這一結果解釋了在AMOVA中種群間的顯著性接近于0.05,可能是受到元江元陽種群組的影響。盡管其他種群間的遺傳分化不明顯,但元江元陽種群組和其他種群存在明顯遺傳分化,具有明顯的種群遺傳結構。
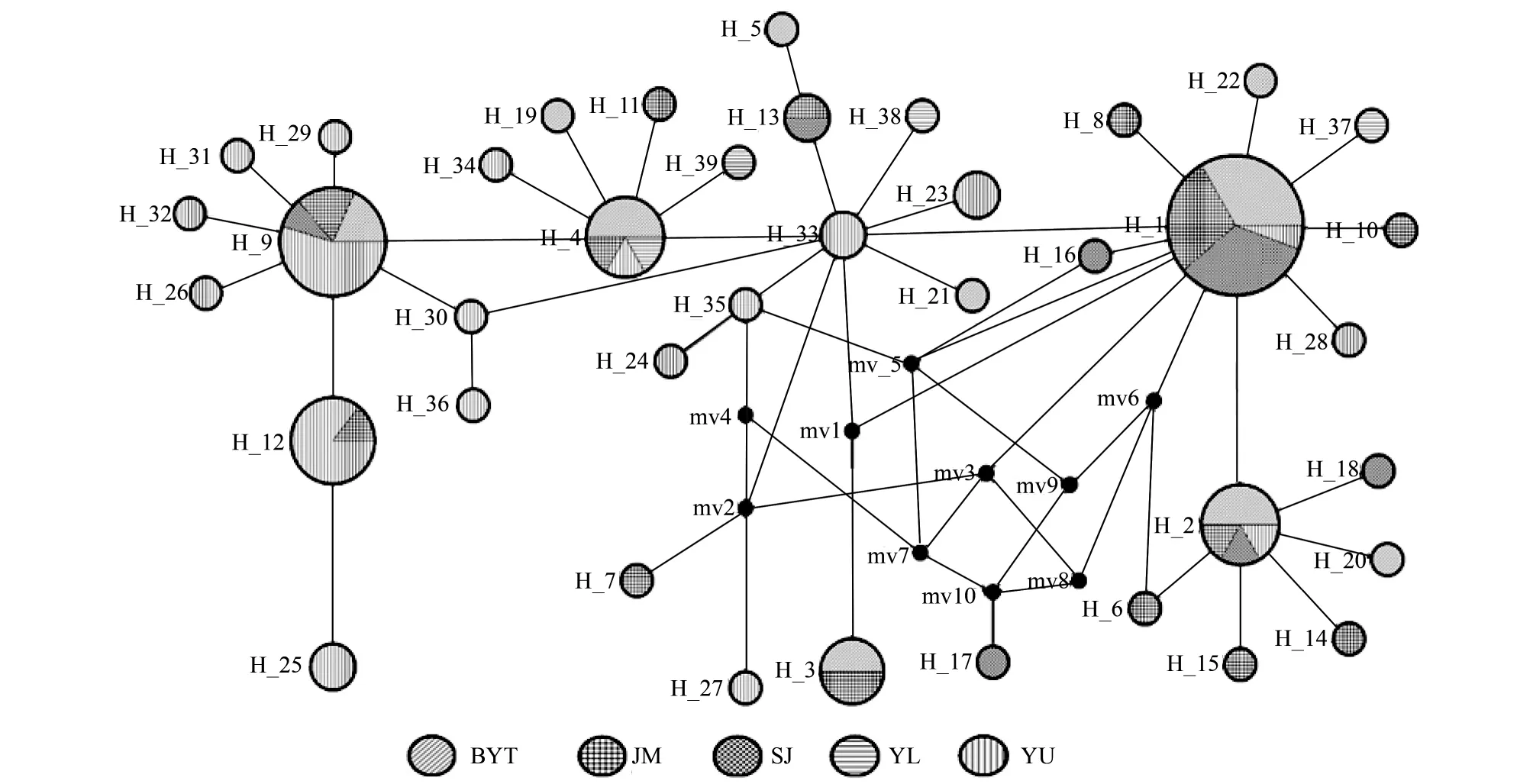
圖5 單倍型的進化網絡圖Fig. 5 Network relationship of haplotypes
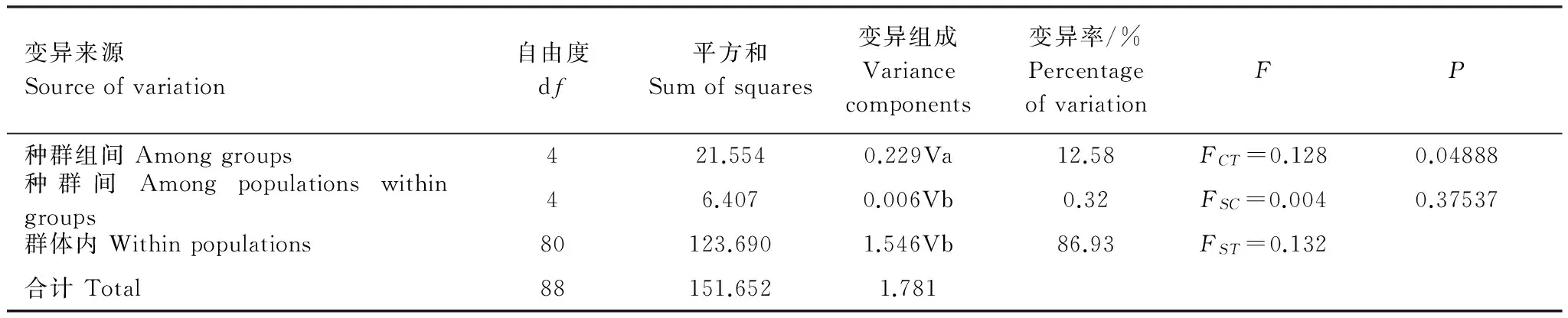
表3 基于線粒體COII的中華按蚊種群分子變異(AMOVA)
2.5 種群歷史動態分析
COII序列的中性測試結果顯示Tajima′sD= -2.252,P< 0.01; Fu and Li′sF*test statistic = -4.892,P<0.02。表明目標序列在進化上遵循中性模型,都為負值,同時歧點分布結果顯示單倍型和堿基差異呈單峰分布(圖6),說明云南地區中華按蚊種群近期經歷過種群擴張事件。
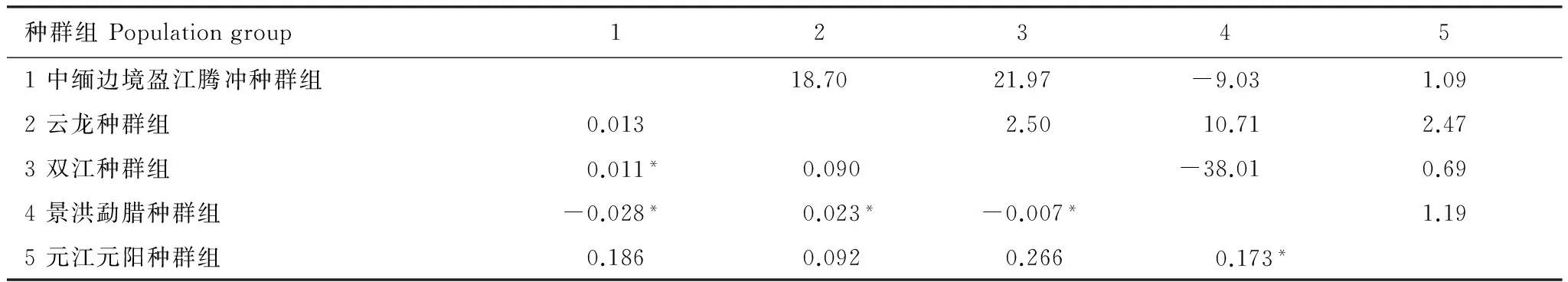
表4 基于COII基因的中華按蚊種群間分化指數(Fst)(對角線之下)與種群基因交流值(Nm)(對角線之上)
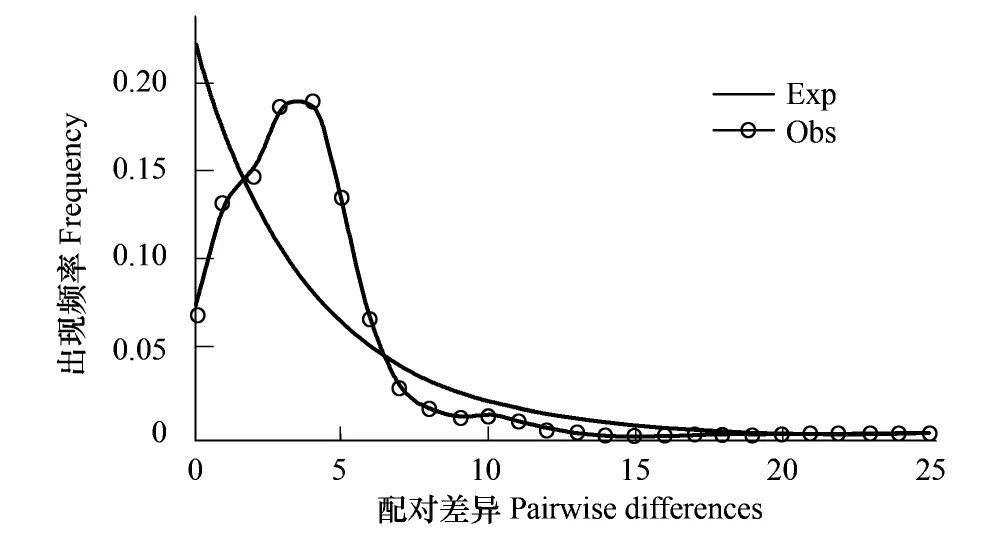
圖6 中華按蚊種群歧點分布檢測Fig. 6 Mismatch-distribution of Anopheles sinensis COII haplotypes
3 討論
3.1 中華按蚊的遺傳多樣性
本研究從云南省9個采樣點廣泛采集、測序和分析了89頭中華按蚊標本。89頭樣本檢測出39個COII單倍型,占總體樣本個數43.8%,各個種群組的單倍型個數百分比都幾乎超過了50%,說明云南中華按蚊具有較高的遺傳性多樣性。雖然不同地區的遺傳多樣性有一定的差別,但總體差別不大,核苷酸多樣性均在0.00406—0.00496之間。中華按蚊受地理分布氣候影響的同時,由于其成蚊偏好吸嗜牛血、人血,幼蟲喜好水稻田等生態特點,是總體遺傳多樣性差別不大的主要原因。單倍型多樣性和核苷酸多樣性最高的是云龍種群組云龍,由于云龍的樣本數較少(僅4個樣本),分析結果存在一定誤差;不考慮云龍遺傳多樣性的情況下,單倍型多樣性和核苷酸多樣性最高的是景洪勐臘種群組景洪勐臘。景洪勐臘地處西雙版納州,屬于熱帶雨林氣候,相對于云南其他地區,常年氣溫較高,氣候濕潤,同時西雙版納州被聯合國教科文組織評為“國際生物圈保護區”,生態環境良好,這些因素是這一地區的中華按蚊遺傳多樣性十分豐富的重要因素。盡管元江元陽種群組的樣本數是最多的,但其單倍型多樣性和核苷酸多樣性都不是最高,這主要是所處的元江元陽地區,常年氣候干燥,氣溫較低,四季交替明顯,并不利于中華按蚊種群遺傳分化。云南地處邊境,常年人口流動大,境外瘧疾的輸入,導致中華按蚊的傳瘧作用尤為突出,所以云南中華按蚊瘧疾傳播的防治工作一直是難點,掌握中華按蚊遺傳多樣性及分化,對中華按蚊及瘧疾控制具有重要的作用。
3.2 種群遺傳結構
本研究基于mtDNA的COII序列建樹分析顯示(圖4),云南中華按蚊各個種群的單倍型呈現出一種混雜的分布格局,沒有檢測到與采樣點嚴格對應的分支,沒有形成明顯的系統地理格局,這與馬雅軍等研究的結論一致。馬雅軍認為中國的中華按蚊種群的分化與其地理距離或者障礙(地理隔離)沒有關系[12],形成這種結果的原因主要是我國中華按蚊易變的有效種群大小和其他一些因素,比如殺蟲劑和對抗瘧疾的免疫基因等都可能影響到種群的分化。而單倍型網絡結構圖卻顯示(圖5),主要以單倍型H1、H9、H4、H33和H2為中心呈星狀分布。盡管大部分種群單倍型沒有按采樣地形成各自的分支,但元江元陽種群組的單倍型分布存在明顯的地理分布,這說明馬雅軍的這一觀點對云南大部分中華按蚊地理分布相符合,但在個別地區仍有差別。
為了進一步了解其地理格局和種群結構,把9個采樣點的樣本按地理分布劃分為5個種群組,AMOVA分析顯示種群組間遺傳變異為12.58%,達到顯著水平(P=0.04888)(表3),說明地理種群組間具有明顯種群遺傳結構。通過Nm和Fst分析表明,云南中華按蚊大部分種群間遺傳分化較小,種群間存在廣泛的基因交流,但元江元陽種群組除了和云龍種群組有基因交流外和其他種群間都缺乏明顯的交流,和其他種群存在明顯分化現象。從理論上講中華按蚊體型較小不適宜長距離遷飛,受限于自身的擴散能力,一定程度的隔離即可顯著限制種群的基因交流,從而導致種群間形成明顯的遺傳分化,但群體分化程度主要是對所在的生態條件相適應的結果,如果生態條件所產生的環境作用強度和方向大體相同,則各個分布區內的種群在遺傳上將難以形成顯著的分化[22]。同時一些自然因素(如風力,河流流動)和人類活動也可能使遠距離的種群間產生基因交流。哀牢山是云南南北走向的大山,成為云貴高原和橫斷山脈兩大地貌的分界線,它的出現使云南呈現東西不同地理氣候,西南部地勢較低,常年降水較多,氣候濕潤,而東部高原氣候雨水少,干燥,常年平均氣溫較低,本次涉及的采樣點元江、元陽位于云南中南部緊鄰哀牢山南端,導致元江元陽種群組和其他種群組分別處于兩種不同的氣候區,同時哀牢山的存在也阻斷了元江元陽種群組和其他種群組的基因交流,導致元江元陽種群組的中華按蚊朝著不同的方向進化,使元江元陽種群組和其他種群組間存在明顯遺傳差異。而其他4個種群間遺傳分化較小主要是因為他們處于同一氣候環境中,加上水稻田是中華按蚊主要的生活環境,而在云南省水稻田分布廣泛,同時現行的水稻殺蟲劑也都主要是毒死蜱、敵百蟲、敵敵畏等常用殺蟲劑,中華按蚊的生活環境和環境壓力大體一致,導致這些地區種群并沒有形成明顯的分化特征。同時瀾滄江是橫跨云南南北處于云南西部的一條大江,本研究所涉及的采樣點除了元江元陽種群組外其他種群都分布在其周圍,瀾滄江水流流動在一定程度上也促進了這些地區中華按蚊的基因交流。
3.3 種群歷史動態
Tajima′D中性檢測值在一定程度內可以反應種群的歷史動態,一個負D值表明存在凈化選擇或群體中有輕微分離的有害突變,也可能是由于群體的擴張引起的。正D值可解釋為平衡選擇將突變保持在平衡頻率,也有可能是一個群體收縮導致的[23]。本研究中性檢測結果均為負值并且顯著,說明整體種群中存在許多的低頻率等位基因突變,歧點分布為明顯單峰分布,單倍型網絡圖反應種群經歷過種群擴張,說明云南地區中華按蚊種群在歷史上經歷過復雜的種群擴張。同時從整個種群平均單倍型偏高,但核苷酸多樣性偏低來看在種群發生擴張的初期云南的中華按蚊有效種群規模較小,后來經歷了迅速的擴張。
引起種群擴張變化的主要因素是氣候變化和地質運動,云南省地質十分復雜,在歷史上也發生過多次地質變遷,特別是元古代的震旦紀到中生代的白堊紀晚期,云南經歷了無數次不同的地殼運動,在第三紀始新世由于一系列的變化,一些類群的重新分布后在新的環境里得到繁衍和發展[24],中華按蚊種群可能在這個時期在適應了新的環境后發生了迅速擴張事件。到了第四紀喜馬拉雅運動時期,地面大規模隆起,哀牢山的形成,阻斷了元江元陽地區中華按蚊與云南其他地區種群的交流,從而逐漸形成了現在這種分布格局。
[1] Harrison B A, Scanlon J E. The subgenusAnophelesin Thailand (Diptera: Culicidae). Contributions of the American Entomological Institute, 1975, 12(1): 1- 307.
[2] Tanaka K, Mizusawa K, Saugstad E S. A revision of the adult and larval mosquitoes of Japan (Including the Ryukyu Archipelago and the Ogasawara Island) and Korea (Diptera: Culicidae). Contributions of the American Entomological Institute, 1979, 16: 1-987.
[3] Rueda L M, Iwakami M, O′guinn M, Mogi M, Prendergast B F, Miyagi I, Toma T, Pecor J S, Wilkerson R C. Habitats and distribution ofAnophelessinensisand associatedAnopheleshyrcanusgroup in Japan. Journal of the American Mosquito Control Association, 2005, 21(4): 458- 463.
[4] 陸寶麟. 中國動物志, 昆蟲綱, 第九卷, 雙翅目, 蚊科(下). 北京: 科學出版社, 1997: 12- 38.
[5] 周水森, 王漪, 房文, 湯林華. 2008年全國瘧疾形勢. 中國寄生蟲學與寄生蟲病雜志, 2009, 28(6): 455- 457.
[6] Chow C Y. Bionomics of malaria vectors in the Western Pacific region. Southeast Asian Journal of Tropical Medicine and Public Health, 1970, 1(1): 40- 57.
[7] Lee W J, Klein T A, Kim H C, Choi Y M, Yoon S H, Chang K S, Chong S T, Lee I Y, Jones J W, Jacobs J S, Sattabongkot J, Park J S.Anopheleskleini,Anophelespullus,andAnophelessinensis: potential vector of Plasmodium vivax in the Republic of Korea. Journal of Medical Entomology, 2007, 44(6): 1086- 1090.
[8] Ree H I, Hwang U W, Lee I Y, Kim T E. Daily survival and human blood index ofAnophelessinensis, the vector species of malaria in Korea. Journal of American Mosquito Control Association, 2001, 17(1): 67- 72.
[9] Coleman R E, Kiattibut C, Sattabongkot J, Ryan J, Burkett D A, Kim H C, Klein T A. Evaluation of anopheline mosquitoes (Diptera: Culicidae) from the republic of Korea for plasmodium vivax circumsporozoite protein. Journal of Medical Entomology, 2002, 39(1): 244- 247.
[10] 錢會霖, 湯林華, 程義亮, 楊寶金. 中華按蚊為唯一傳瘧媒介地區瘧疾傳播潛勢的初步估算, 中國寄生蟲學與寄生蟲病雜志, 1994, 12(4): 265- 267.
[11] Merrell D J. Ecological Genetics. London: Longman, 1981: 453- 491.
[12] Ma Y J, Yang M N, Fan Y, Wu J, Ma Y, Xu J N. Population structure of the malaria vectorAnophelessinensis(Diptera: Culicidae) in China: Two gene pools inferred by Microsatellites. PLoS ONE, 2011, 6(7): e22219.
[13] 董學書,周紅寧,龔正達. 云南蚊類志(上卷). 昆明: 云南科技出版社, 2009: 144- 146。
[14] Joshi D, Park, M H, Saeung A, Choochote W, Min G S. Multiplex assay to identify Korean vectors of malaria. Molecular Ecology Resources, 2010, 10(4): 748- 750.
[15] Chen B, Pedro P M, Harbach R E, Somboon P, Walton C, Butlin R K. Mtiochondrial DNA variation in the malaria vectorAnophelesminimusacross China, Thailand and Vietnam: evolutionary hypothesis, population structure and population history. Heredity, 2011, 106(2): 241- 252.
[16] Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetic analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 2011, 28(10): 2731- 2739.
[17] Librado P, Rozas J. DnaSP v5: a sotfware for comprehensive analysis of DNA polymorphism data. Bioinformatics, 2009, 25(11): 1451- 1452.
[18] Nei M. Evolution of human races at the gene level // Bonne-Tamir B, Cohen T, Goodman R M. Human Genetics, part A: The Unfolding Genome. New York: Alan R. Liss, 1982: 167- 181.
[19] Roger A R, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Molecular Biology and Evolution, 1992, 9(3): 552- 269.
[20] Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evolutionary Bioinformatics, 2005, 1: 47- 50.
[21] Bandelt H J, Forster P, Rohl A. Median-joining network for inferring intraspecific phylogenies. Molecular Biology and Evolution, 1999, 16(1): 37- 48.
[22] Kelley S T, Farrell B D, Mitton J B. Effects of specialization on genetic differentiation in sister species of bark beetles. Heredity, 2000, 84(2): 218- 227.
[23] 梁日霞, 王振營, 何康來, 叢斌, 李菁. 基因線粒體COII基因序列的雙斑長跗螢葉甲中國北方地理種群的遺傳多樣性研究. 昆蟲學報, 2011, 54(7): 828- 837.
[24] 朱世模, 黃復生. 地質變遷與云南白蟻的變化. 動物學研究, 1989, 10(1): 1- 8.
Genetic variation and population structure ofAnophelessinensis(Diptera:Culicidae) in Yunnan
YANG Feilong, LI Xudong, YAN Zhentian, FU Wenbo, CHEN Bin*
InstituteofEntomologyandMolecularBiology,CollegeofLifeSciences,ChongqingNormalUniversity,Chongqing401331,China
Anophelessinensis(Diptera: Culicidae) is an important malarial vector in Yunnan Province, China. In order to explore the genetic variation and population structure among different populations of the species, we sequenced and analyzed mitochondrialCOIIsequences of 89An.sinensissamples. These samples were collected from nine localities in Yunnan, and were classified into five population groups based in different geographical locations. The results showed thatAn.sinensishad high genetic diversity in Yunnan with the haplotype diversity (h) and nucleotide diversity (π) being 0.933 and 0.00406, respectively. Fifty-one variations were found, occupying 6.9% of the total 739 bp of theCOIIsequence. Thirty-nine haplotypes were identified, making up 43.8% of 89 samples, with H1 and H9 being the most widespread, occupying 20.2% and 12.4% of the total haplotypes, respectively. The Jinghong-Mengla population group (JM) has the highest haplotype and genetic diversity, followed by the Yunlong population group (YL). The tropical rainforest region in Xishuangbanna, which is characterized by high temperature, humidity, and a primary ecological environment, might contribute the most to the high genetic diversity of the JM. Phylogenetic analysis did not reveal a significant relationship between the haplotypes and geographical locations. The haplotype network diagram did not reveal a relative geographical pattern of haplotype distribution, with the exception of the Yuanjiang-Yuanyang population group (YU) of haplotypes that showed obvious characteristics associated with regional distribution. Haplotypes were mainly distributed around the haplotypes H1, H9, H4, H33, and H2. Analysis of molecular variance (AMOVA) indicated the existence of population structure among population groups. No obviousFstandNmvalues were observed among four population groups (ZYT, YL, SJ, and JM) from seven sampling localities, which might indicate that there are no obvious genetic differences and that gene flow has occurred among these four population groups. However, the population group YU lacked any significant genetic exchange with other populations leading to clear genetic differentiation. For the former, this might result from the wide distribution of paddy fields, which is the predominant habitat ofAn.sinensis. For the latter, the Mountain Ailaoshan barrier, a region with a unique geography and climate, might result in genetic separation. Yuanjiang and Yuanyang for YU are located north-south from Mountain Ailaoshan so that the group separated from the other four groups into two different climate environments. Mismatch distribution was unimodal and the Neutral Test values were significantly negative. These might indicate that a complicated population expansion occurred in theAn.sinensispopulations in Yunnan. The high percentage of haplotypes and low percentage of nucleotide diversity also supports the recent population expansion of the species in Yunnan. The population expansion might be relative to the geological changes in Yunnan, especially in the period from the late Proterozoic sinian to the Mesozoic cretaceous. During this period, Yunnan experienced a number of crustal movements, and some groups of mosquitoes survived following the changes, and adapted and expanded into new environments. This study provides information about genetic diversity and population characteristics, and is of importance for the control ofAn.sinensisand malaria in Yunnan.
Yunnan;Anophelessinensis; mtDNACOII; genetic variation; population structure
“兩江學者”計劃專項經費;美國國立衛生研究院NIH項目(R01AI095184);國家自然科學基金(31071968,31372265);重慶市科技攻關重點項目(CSTC2012GG-YYJSB80002)
2013- 11- 17;
2014- 08- 20
10.5846/stxb201311172746
*通訊作者Corresponding author.E-mail: bin.chen@cqnu.edu.cn
楊飛龍,李旭東,閆振天,付文博,陳斌.云南中華按蚊的遺傳變異和種群結構.生態學報,2015,35(16):5449- 5457.
Yang F L, Li X D, Yan Z T, Fu W B, Chen B.Genetic variation and population structure ofAnophelessinensis(Diptera: Culicidae) in Yunnan.Acta Ecologica Sinica,2015,35(16):5449- 5457.
