15例兒童炎癥性肌纖維母細胞性腫瘤的臨床及病理分析
2015-03-15 03:34:08呂孟興
檢驗醫學與臨床 2015年2期
呂孟興,高 燕,周 軍
(昆明市兒童醫院 650028)
?
·臨床研究·
15例兒童炎癥性肌纖維母細胞性腫瘤的臨床及病理分析
呂孟興,高 燕,周 軍
(昆明市兒童醫院 650028)
目的 探討兒童炎癥性肌纖維母細胞性腫瘤(IMT)的臨床特點、病理組織學特征及鑒別診斷。方法 回顧性分析2006~2014年15例兒童IMT的臨床資料,進行組織形態學分析及免疫表型檢測。結果 15例IMT術后隨訪時間為3~60個月,其中10例無瘤生存,3例術后半年到1年內復發,1例發生惡變。1例失訪。免疫組化結果:梭形細胞胞質內波形蛋白、平滑肌肌動蛋白、肌特異性肌動蛋白、結蛋白陽性率分別為100%(15/15)、87%(13/15)、80%(12/15)、47%(7/15);間變型淋巴瘤激酶陽性率為40%(6/15);Ki-67陽性超過15%1例,Ki-67陽性10%2例,Ki-67陽性小于5%12例。CD117、S-100、CD34(內皮細胞標記)、肌調節蛋白、肌漿蛋白、肌紅蛋白均為陰性。結論 兒童IMT是具有惡變傾向的間葉性腫瘤,具有一定的復發率。手術徹底切除仍為目前首選的治療方法,放療和化療的作用有待進一步探討。
炎癥性肌纖維母細胞性腫瘤; 兒童; 免疫組織化學; 鑒別診斷
炎癥性肌纖維母細胞性腫瘤(IMT)是一種中間性軟組織腫瘤,由肌纖維母細胞性梭形細胞和漿細胞、淋巴細胞、嗜酸性粒細胞等炎癥細胞構成。同義詞有漿細胞肉芽腫、漿細胞假瘤、炎癥性肌纖維母細胞性增生、網膜腸系膜性黏液樣錯構瘤、炎性假瘤。其局部復發、浸潤性生長方式及染色體異常的特點,支持這一病變為真性腫瘤。
1 資料與方法
1.1 一般資料 收集昆明市兒童醫院2006~2014年明確診斷為IMT的病例15例,其中男8例,女7例。年齡13 d至12歲,平均8歲,中位年齡5歲。發病部位為肺部3例,腸系膜3例,回盲部2例,結腸2例,盆腔2例,腎臟1例,腹膜后1例,頭皮1例。
1.2 儀器與試劑 免疫組織化學染色試劑一抗、二抗、DAB顯色系統均購自Dako公司。
1.3 方法 標本均經10%中性甲醛固定,石蠟包埋,3 μm厚切片,常規制片。免疫組織化學采用EnVision兩步法。
2 結 果
2.1 腫瘤大體情況 15例送檢標本最大者為8.5 cm×5.8 cm×4.0 cm,最小者為3.0 cm×2.0 cm×1.2 cm,界限清楚或多結節狀,質韌,切面灰白色或灰黃色,呈旋渦狀、編織狀或黏液樣。少數有局灶出血、壞死或鈣化。
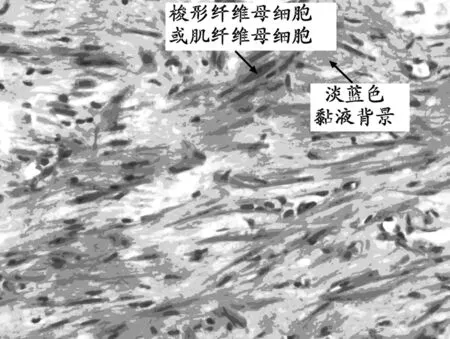
注:瘤細胞梭形或短梭形,稀疏排列,混有炎細胞,背景水腫
性黏液樣。
圖1 腫瘤細胞HE染色(×400)
2.2 鏡下觀察 腫瘤細胞均為梭形或短梭形,稀疏或緊密排列,核分裂少見;混有炎癥細胞,背景為水腫性黏液樣(圖1)。部分病例中可見神經節樣肌纖維母細胞核大泡狀,核仁嗜酸性,細胞質豐富(圖2)。部分病例腫瘤膠原化明顯,細胞成分少,炎細胞少,可有鈣化和骨化,似瘢痕或硬化型纖維瘤病。
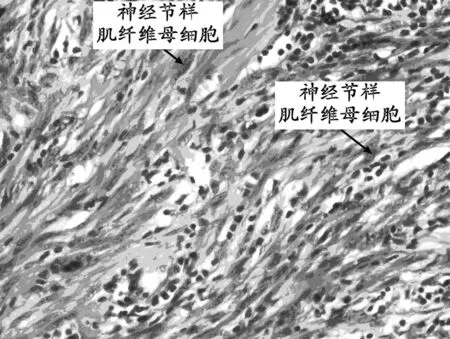
注:神經節樣肌纖維母細胞核大泡狀,核仁嗜酸性,胞質豐富。
圖2 神經節樣肌纖維母細胞HE染色(×400)
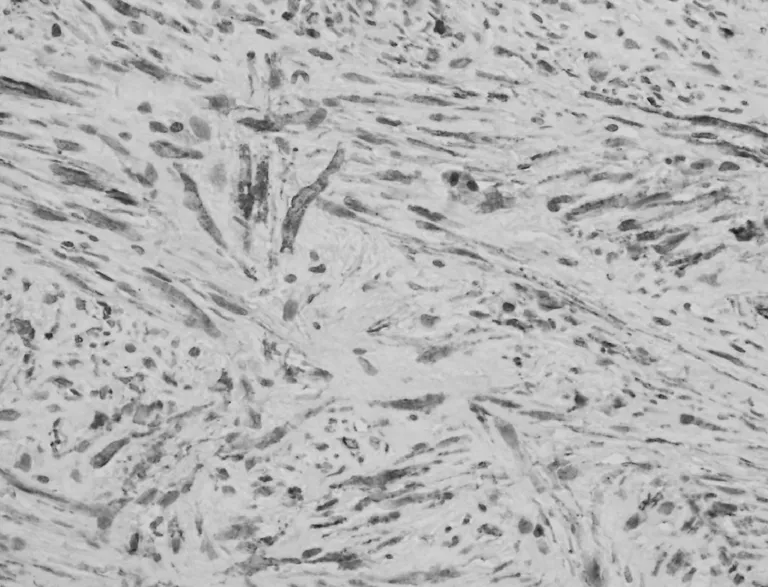
圖3 梭形細胞MSA染色呈塊狀或彌漫的陽性(EnVision兩步法,×400)
2.3 免疫表型 梭形細胞胞質內波形蛋白(vimentin)、平滑肌肌動蛋白(SMA)、肌特異性肌動蛋白(MSA)、結蛋白(Desmin)陽性率分別為100%(15/15)、87%(13/15)、80%(12/15)、47%(7/15);間變型淋巴瘤激酶(ALK)陽性率為40%(6/15);Ki-67陽性超過15%1例,Ki-67陽性10% 2例,Ki-67陽性小于5%12例。CD117、S-100、CD34(內皮細胞標記)、MyoD1(肌調節蛋白)、肌漿蛋白(myogenin)、肌紅蛋白(myoglobin)均為陰性。
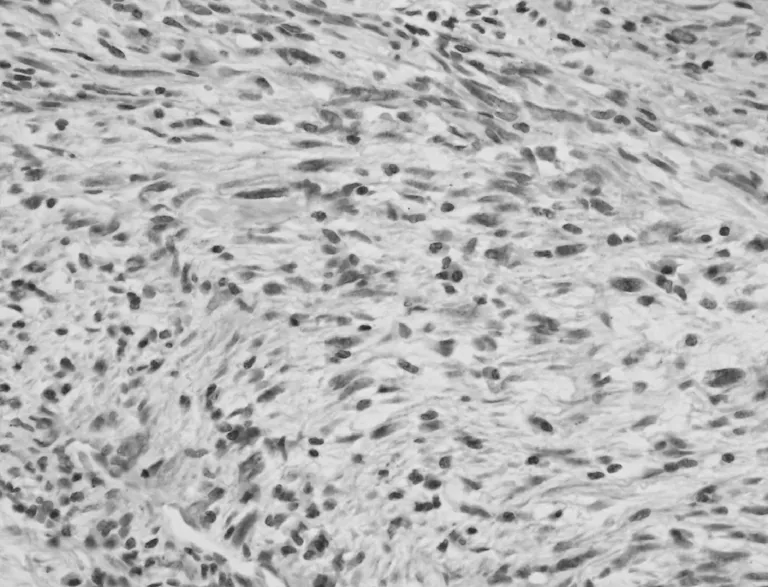
圖4 梭形細胞ALK染色陽性表達(EnVision兩步法,×400)
2.3 免疫表型 梭形細胞胞質內波形蛋白(vimentin)、平滑肌肌動蛋白(SMA)、肌特異性肌動蛋白(MSA)、結蛋白(Desmin)陽性率分別為100%(15/15)、87%(13/15)、80%(12/15)、47%(7/15);間變型淋巴瘤激酶(ALK)陽性率為40%(6/15);Ki-67陽性超過15%1例,Ki-67陽性10%2例,Ki-67陽性小于5%12例。CD117、S-100、CD34(內皮細胞標記)、MyoD1(肌調節蛋白)、肌漿蛋白(myogenin)、肌紅蛋白(myoglobin)均為陰性。
2.4 隨訪 14例獲得隨訪資料,隨訪時間3~60個月。其中10例恢復良好,無復發轉移,腫瘤位于肺、腎、結腸及腸系膜。3例術后半年到1年內復發,腫瘤位于回盲部、腹膜后。1例發生惡變,腫瘤位于盆腔。1例失訪。
3 討 論
IMT最早由Brunn等于1939年報道了2例肺梭形細胞良性腫瘤,認為是一種炎癥性瘤樣病變。Umiker等在1954年提出肺內的這種梭形細胞增生是炎癥后腫瘤(post inflammatory tumors)的觀點。不久炎癥后腫瘤這一名稱逐漸過渡成為易于理解和廣泛應用的名稱“炎性假瘤”。最近,WHO軟組織腫瘤分類中將其歸為纖維母細胞/肌纖維母細胞腫瘤、中間性、偶有轉移型。將IMT定義為由分化的肌纖維母細胞性梭形細胞組成的,常伴大量漿細胞和(或)淋巴細胞的一種腫瘤。IMT是一種少見的間葉性腫瘤,其病因與發病機制目前尚不清楚,部分病例發生在手術、創傷或炎性反應以后,提示IMT可能是人體對損傷的一種異常或過度的反應。有文獻報道1例病例起病前曾行患側附耳切除手術,術后反復感染經久不愈,推測創傷等原因引起的炎癥反應刺激與IMT發生關系密切[1]。IMT多發生于兒童及年輕人,平均年齡10歲,中位數為9歲。最常發生的部位是肺部[2],肺外的部位包括腸系膜、大網膜、腹膜后、膀胱、中樞神經系統、軟組織、胃腸道、肝、腎、脾、胰等[3-5]。發生部位廣泛,在不同的部位表現多樣,肺部IMT以反復呼吸道感染、咳嗽、發熱,常以支氣管肺炎入院,腹腔內IMT可出現腹痛、腹脹,腹部包塊等。膀胱內IMT多以血尿為主要癥狀。發生于回腸的IMT可引起回結型腸套。
3.1 診斷與鑒別診斷 IMT臨床及影像學缺乏特異性表現,確診依賴病理學。IMT光鏡下主要由增生的肌纖維母細胞、纖維母細胞、膠原纖維和炎性細胞組成。纖維母細胞/肌纖維母細胞大部分呈梭形或短梭形,胞質嗜酸性,核卵圓形,居中,可見核仁,輕度異型性,核分裂象少。Coffin等[6]提出3種組織學類型:(1)黏液樣/血管型。以黏液、血管、炎癥細胞為主,可類似結節性筋膜炎和橫紋肌肉瘤。(2)梭形細胞密集型。以梭形細胞為主,間雜炎細胞,類似纖維組織細胞瘤。(3)纖維型。以膠原纖維為主類似瘢痕或硬化性纖維瘤少部分出現點狀或大片狀的鈣化和化生骨。本組病例中以梭形細胞密集型多見,為12例,黏液血管型2例,纖維型1例。IMT在病理形態上應與一些良、惡性的梭形細胞腫瘤病變進行鑒別,常見的鑒別診斷如下:
3.1.1 結節性筋膜炎 多發生于皮下或淺筋膜的結節狀肌纖維母細胞性增生,細胞為梭形,呈不規則的短束狀或交織狀排列,間質疏松,黏液樣,可見外滲的紅細胞和散在的淋巴細胞,部分病例可見微囊性腔隙,通常不見漿細胞。ALK陰性。
3.1.2 炎癥性惡性纖維組織細胞瘤 多發生于成年人,腫瘤細胞呈多形性,異型性明顯,有多量的黃色瘤細胞核炎癥細胞,核分裂活躍,可見病理性核分裂象。免疫組織化學不表達肌源性標記。
3.1.3 平滑肌肉瘤 多發生于中老年患者,絕大多數病例發生于盆腔腹膜后或腹腔,腫瘤細胞主要由平行束狀或交織束狀排列的嗜伊紅色梭形細胞組成。核兩端平鈍或呈雪茄樣,部分瘤細胞核的一端見空泡,常形成凹陷性壓跡。病理性核分裂像多見,一般無炎性細胞彌漫浸潤。70%~80%病例表達結蛋白。
3.1.4 纖維瘤病 多發生于新生兒和嬰幼兒,有明顯的區帶現象,由淡染的周邊區和深染的中心區組成,瘤細胞為結節狀或短束狀排列的胖梭形纖維母細胞組成,其間有數量不等的膠原纖維。少見炎細胞浸潤。
3.1.5 胃腸道、網膜及腹膜后間質瘤 好發于40~70歲,較少發生于兒童和青少年。腫瘤細胞呈梭形或短梭形,多呈交織的短條束狀或漩渦狀排列,部分病例核端可見空泡或呈兩端平鈍的雪茄樣。一般無炎細胞彌漫浸潤。CD117和CD34陽性。
3.2 治療與預后 目前治療IMT的方法有根治性切除、激素治療、放療和化療。大部分病例預后較好,部分IMT有復發和遠處轉移,少數還可發生惡變。肺外IMT復發率約為20%~25%,轉移率小于5%[7]。最常見轉移部位是肺和腦,其次是肝和骨。轉移最常發生于診斷后1年內,偶有發生于手術切除后9年的報道,術后長期隨訪很重要[8]。有文獻報道對于IMT放化療效果不明確,但對于侵襲性強的IMT推薦輔助放化療[9]。目前,酪氨酸酶抑制劑(erizotinib)用于ALK陽性的IMT患者有個別報道,并取得了一定的效果。ALK成為潛在的分子治療靶點,受體酪氨酸激酶抑制劑靶向治療對多次復發或因發生部位無法完全手術切除的患者提供了新的選擇[10]。Ong等[11]提出Ki-67和ALK陽性表達、腫瘤切緣陽性及腫瘤大小與預后和局部復發關系密切,尤其是Ki-67和ALK陽性同時存在時,術后應給予化療或局部放療。鑒于IMT有復發、轉移甚至惡變的可能,有必要進行長期追蹤隨訪。
[1]劉艷麗,劉良發.頭頸部炎性肌纖維母細胞瘤臨床分析[J].中華耳鼻咽喉頭頸外科雜志,2014,49(1):35-38.
[2]Guan Y,Chen G,Zhang W,et al.Com-puted tomography appear ance of inflammatory myofibroblastic tumor in the mediastinum[J].J Comput Assist Tomogr,2012,36 (6):654-658.
[3]Whitehead MT,Grimm J,Nelson MD.Case 185:inflammatory myofibroblastic tumor[J].Radiology,2012,264(3):912-916.
[4]Jeon YK,Chang KH,Suh YL,et al.Inflammatory myofibroblastic tumor of the central nervous system:clinicopathologic analysis of 10cases[J].J Neuropathol Exp Neurol,2005,64(3):254-259.
[5]Meng X,Wang R.Inflammatory myofibroblastic tumor oreurs in the mediastinum[J].J Cancer Res Ther,2013,9(4):721-723.
[6]Coffin CM,Watterson J,Priest JR,et al.Extrapulmonary inflame matory myofibroblastic tumor inflammatory pseudotumor.A clinicopathologic and immunohistochemical study of 84 cases[J].Am J Surg Pathol,1995,19(8):859-872.
[7]Stelow EB,Shami VM,Moskaluk CA,et al.Esophageal inflammarory myofibroblastic tumor sampled by EUS-FNA[J].Gastrointestinal Endoscopy,2010,72(1):209-210.
[8]Yeh YS,Ma CJ,Yang SF,et al.Inflammatory myofibroblastic tumor of the lleum causing an unusual Ileocecal intussusception[J].Fooyin J Health Sci,2010,2(1):36-39.
[9]Coffin CM,Hornick JL,Fletcher CD.Inflammarory myofibroblastin tumor:comparison of clinicopatholgic,histologic,and immohistochemical features including ALK expression in atypical and aggressive cases[J].Am J Surg Pathol,2007,31(4):509-520.
[10]Butrynski JE,D′Adamo DR,Hornick JL,et al.Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor[J].N Engl J Med,2010,363(18):1727-1733.
[11]Ong HS,Ji T,Zhang CP,et al.Head and neck inflammatory myofibroblastic tumor(IMT):evaluation of clinicopathologic and prognostic features[J].Oral Oncotogy,2012,48(2):141-148.
10.3969/j.issn.1672-9455.2015.02.036
A
1672-9455(2015)02-0223-02
2014-06-09
2014-11-09)
