苯胺及摻雜態苯胺低聚物結構的密度泛函理論研究
2015-03-23 11:56:23薛嚴冰于婧怡唐禎安
原子與分子物理學報 2015年2期
關鍵詞:結構
薛嚴冰, 于婧怡, 許 芝, 唐禎安
(1.大連交通大學電氣信息學院, 大連 116028; 2.大連理工大學電子科學與技術學院, 大連 116023)
苯胺及摻雜態苯胺低聚物結構的密度泛函理論研究
薛嚴冰1, 于婧怡1, 許 芝1, 唐禎安2
(1.大連交通大學電氣信息學院, 大連 116028; 2.大連理工大學電子科學與技術學院, 大連 116023)
采用基于密度泛函理論的計算方法,研究了苯胺低聚物及鹽酸、對甲苯磺酸摻雜苯胺低聚物的幾何結構和電子結構.結果表明,質子酸摻雜使苯胺低聚物分子鏈上醌環中C-C單、雙鍵交替的性質被削弱,同時鏈間C=N鍵長明顯增大.摻雜位上鏈間C-N-C鍵角增大,相鄰環間的扭轉角減小,分子鏈的共面性有所改善.與鹽酸摻雜對比,對甲苯磺酸摻雜更利于電子從環內向鏈間轉移,理論上可更好地改善聚苯胺材料的導電性能.
苯胺低聚物; 密度泛函法; 幾何結構; 電子結構
1 引 言
聚苯胺(Polyaniline,PANI)是一種典型的導電高分子材料,具有原料易得、合成簡單、良好的環境穩定性、導電性、光電性、熱電性等諸多優點,近幾年已引起研究者的廣泛關注.有關其物理、化學性質、制備方法及應用等的實驗研究已取得一定進展[1-4],但從理論計算角度出發,著眼于聚苯胺分子的微觀結構及導電機理的研究還相對較少.
Lim S. L.等[5]是最早采用量子計算方法研究聚苯胺材料特性的.他們分別采用基于第一性原理(ab initio)和密度泛函理論(DFT)計算的方法研究了幾種中性苯胺低聚體的幾何和電子結構,研究結果表明用DFT方法得到的計算結果同由X射線衍射、紫外光吸收譜和X射線光電子能譜等實驗觀察到的數據很好地保持一致.此后陸續有采用第一性原理和(或)DFT計算方法研究苯胺低聚體的成果報道[6-16].Foreman J. P.等[7]用DFT的方法研究了樟腦磺酸(CSA)摻雜的還原結構苯胺低聚體,發現胺和磺酸基團間的氫鍵增加了苯-氮-苯骨架轉移電子密度的能力.Yang G.等[14]分析了鹽酸和樟腦磺酸摻雜的苯胺低聚體的電子結構,發現后一種摻雜較前一種摻雜能引起更多的電荷轉移.Casanovas J.等[15]用DFT方法對聚苯胺低聚體進行研究,他們建立了從甲基到戊基不同的烷基硫酸鹽陰離子,鏈上包含2-6個苯環的低聚體模型,系統研究了摻雜烷基的長度和聚合物鏈模型的大小對計算結果的影響.
由于計算模型、計算方法、計算參數等的不同,上述研究結果并不完全具有可比性.特別是針對摻雜態聚苯胺,已有的研究只涉及到鹽酸和樟腦磺酸摻雜.有研究表明對甲苯磺酸(P-TSA)摻雜可提高聚苯胺材料對某些氣體的響應靈敏度,有望實現其在氣敏材料方面的應用[17],而有關P-TSA摻雜苯胺低聚體的理論計算尚未見有文獻報道.本文采用基于密度泛函理論的計算方法,建立本征聚苯胺低聚體模型及鹽酸和P-TSA摻雜聚苯胺低聚體模型,研究并比較不同酸摻雜態苯胺低聚體的幾何結構和電子結構,提出質子酸摻雜改善聚苯胺導電性能的機理,為有針對性地設計導電聚苯胺材料提供理論依據.
2 模型與計算方法
2.1 理論模型
PANI目前廣為接受的分子鏈模型是1986年MacDiarmid提出的苯式-醌式結構單元共存的模型[18]:
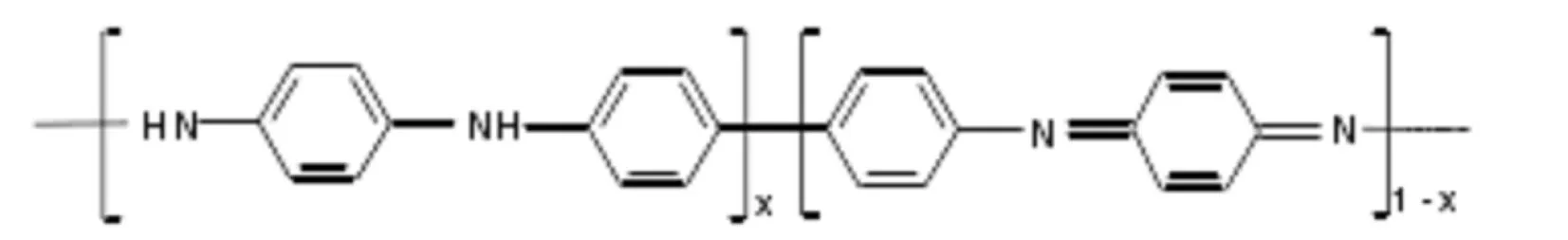
式中x代表PANI的氧化程度,當x=1時為完全還原態的全苯式結構,x=0時為完全氧化態的苯-醌交替結構;而x=0.5為苯-醌比為3:1的半氧化結構.在上述3種可穩定存在的PANI形態中,只有半氧化態PANI能通過摻雜發生從絕緣態到導電態的突變[19],因此本文以半氧化態PANI作為初始計算模型.
2.2 計算方法
采用基于密度泛函的理論計算方法,應用Accelrys 公司Material Studio軟件包中的Dmol3計算模塊進行計算[20].參數設置為:采用廣義梯度近似(GGA),PBE泛函[14],結構優化采用BFGS計算方法,電子設置為全電子方式,基組選擇為極化的雙數值基組(DNP),SCF收斂判據設為中等精度的收斂條件.為加快收斂速度,選用了DIIS算法.先對建好的分子構象進行幾何優化,再進行電子特性計算.
3 結果與討論
3.1 PANI低聚物分子鏈結構的確定
分別建立了包含4、8、12、16個芳環的半氧化態苯胺低聚體模型,研究芳環數對分子鏈幾何結構的影響.幾何參數包括鍵長(環內的C-C鍵長及環間的C-N鍵長)、C-N-C鍵角及相鄰兩環之間的扭轉角.計算結果表明,苯環內C—C單鍵鍵長在0.1395 nm-0.1454 nm之間,C=C雙鍵鍵長在0.1382 nm-0.1415nm之間,單鍵鍵長縮短而雙鍵鍵長拉長,說明形成了共軛長鏈體系[21].芳環數增加對C-C鍵長的影響不明顯,不同鏈長度的C-C平均鍵長基本不變.芳環數增加對C-N鍵長有一定影響,C—N平均長度由4環的0.1384 nm增加至16環的0.1392 nm,C=N平均鍵長也由4環的0.1313 nm增加至16環的0.1323 nm.隨環數增加,C-N-C鍵角平均值有增加趨勢(圖1(a)),相鄰環間扭轉角有減小趨勢(圖1(b)).當環數由12個增加到16個時,鍵角平均值趨于穩定,約為126°,扭轉角平均值亦基本保持恒定,約為41.7°.
上述結果說明,當聚合物包含16個芳環時,體系的結構已趨于穩定,比較接近真實的聚合物片段.考慮到環數近一步增加時,會帶來計算時間的成倍增加,故選擇分子鏈模型為16環聚合物.
3.2 HCl摻雜態苯胺低聚物
圖2是HCl摻雜苯胺低聚物的計算模型.首先建立HCl分子模型并幾何優化,優化后H—Cl鍵長為0.1285 nm,比理論值鍵長0.127 nm略長;鍵能為425.2 KJ/mol,比理論鍵能431 KJ/mol 略小.按照優化后的分子結構,將HCl分子以H原子靠近醌環兩側N原子的方式置于苯胺低聚體鏈上.摻雜方式采用雙極化子摻雜方式[9],即醌環兩側的N原子均為摻雜點.
3.2.1 幾何結構
經優化得到苯胺低聚物的穩定構型.可以發現分子鏈具有規則的扭轉性,同時H—Cl鍵斷開,H質子與亞胺基的氮原子成鍵.HCl摻雜前后苯胺低聚物C-C鍵長的計算結果如圖3(a)所示.摻雜對苯環中C-C鍵長影響不明顯,卻很大程度上改變了醌環中的C-C鍵長值.醌環中C-C單、雙鍵交替的性質被明顯削弱,醌環有被還原為苯環的趨勢.圖3(b)為HCl摻雜前后苯胺低聚物C—N鍵長的計算結果.同摻雜前相比,摻雜位置上C—N單鍵長基本保持不變,而C=N雙鍵鍵長則明顯增加,平均鍵長由摻雜前的0.1323 nm增加至0.1357 nm.說明H質子的加入對醌環上C-N鍵的影響遠大于對苯環上的C-N鍵.
圖4(a)是HCl摻雜前后聚合物的C-N-C鍵角的計算值.經分析,未摻雜點的C-N-C鍵角略微減小,平均值由129.37°減小至128.71°;而摻雜點位置上的C-N-C鍵角明顯的增加,平均值由123.97°增加至129.39°,這一結論和文獻[7,14]的計算結果保持一致.可能的原因是由于氨基N原子同摻入的H質子間形成氫鍵,N的孤對電子轉移到摻雜物和聚苯胺骨架中,使C-N-C鍵角增加.
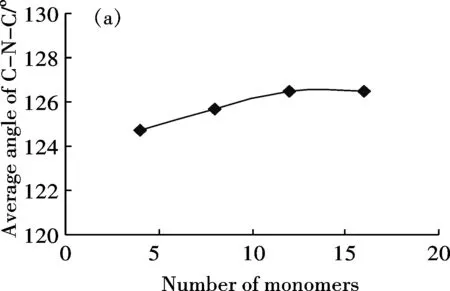
(a)Average angle of C-N-C bond
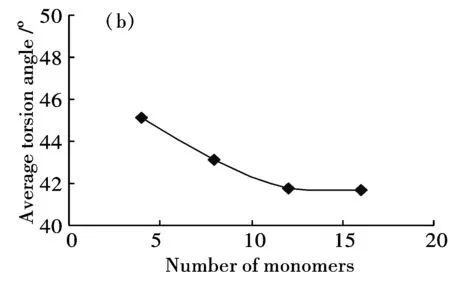
(b)Average torsion angle
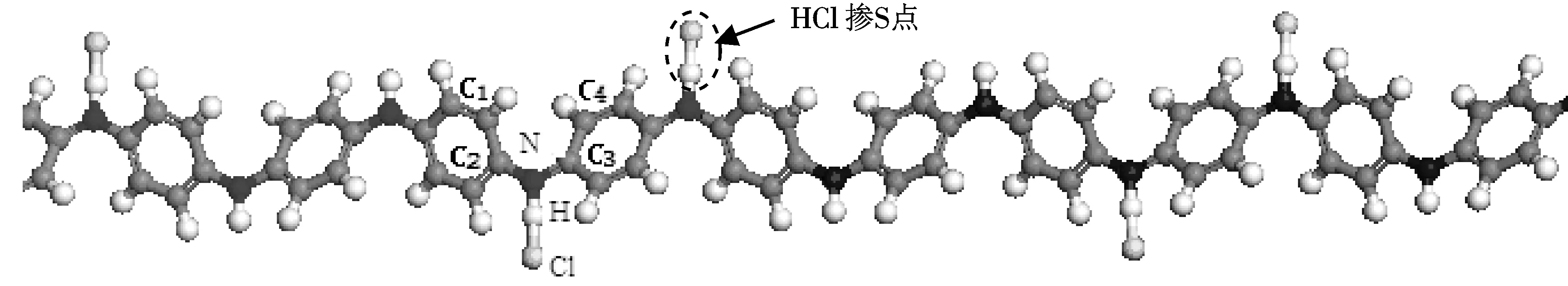
圖 2 HCl摻雜苯胺低聚物的計算模型Fig.2 Calculation models of HCl doped Oligoaniline
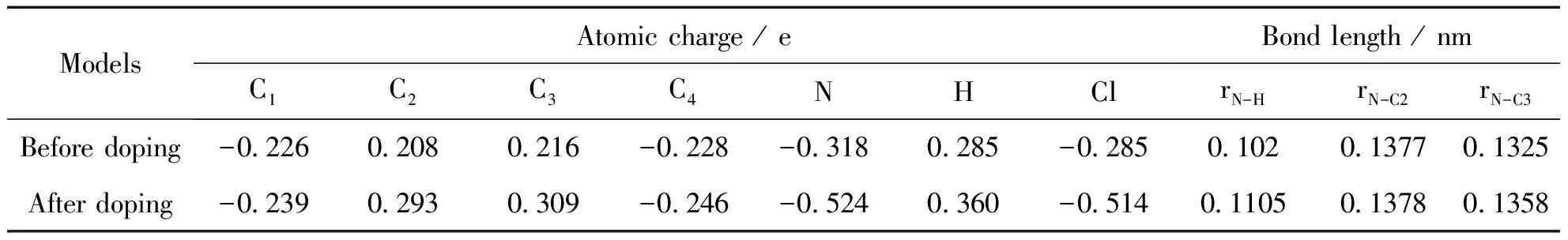
表1 HCl摻雜點附近的原子電荷和鍵長
HCl摻雜前后聚合物相鄰芳環之間扭轉角的計算結果圖4(b).通常情況下,分子平面性越好,分子間的軌道重疊程度越高,其導電能力也越強.可以發現,摻雜位置上相鄰芳環間的扭轉角明顯減小,平均扭轉角由41.57°減小到33.94°.扭轉角代表著N原子的Pz軌道同芳環中π電子的共軛程度,摻雜后相鄰環的共面性有所改善,體系π共軛效果增加,電子易于沿著芳環的骨架進行離域.
3.2.3 電子結構
按照圖2(a)中HCl摻雜點附近的原子標號,表1列出摻雜前后體系內各原子的原子電荷計算結果,表中的數值為8個摻雜點計算結果的平均值.
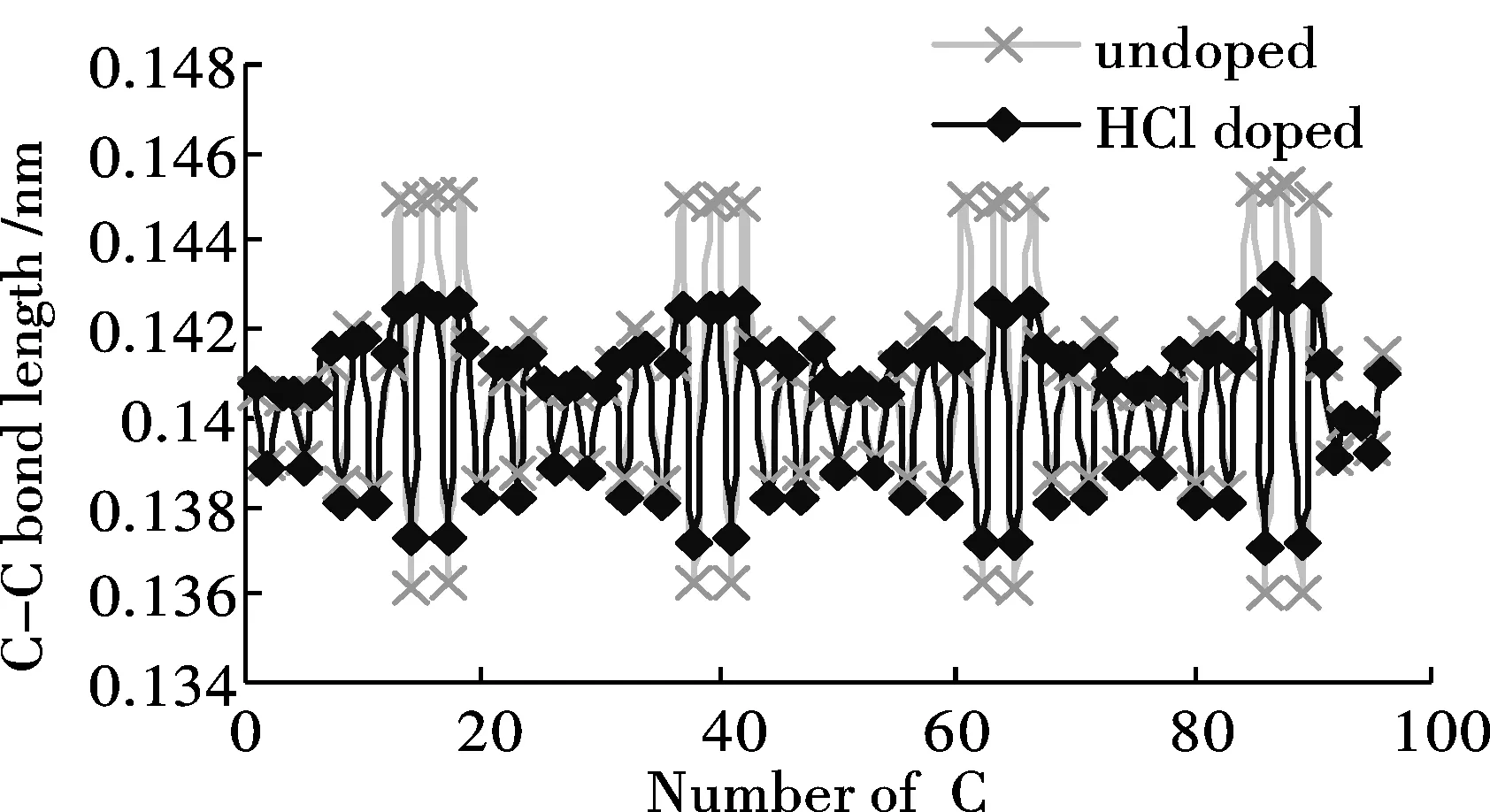
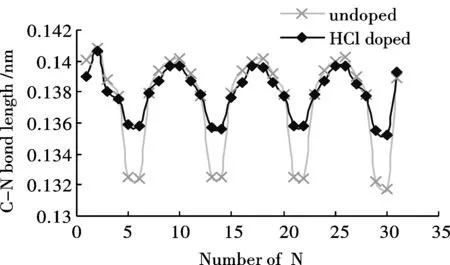
圖 3 HCl摻雜對低聚物鍵長的影響Fig.3 Influence of HCl doping on the bond lengths of Oligoaniline


圖4 HCl摻雜對低聚物鍵角的影響Fig.4 Influence of HCl doping on the bond angles of Oligoaniline
從表中可以看出,摻雜后C2和C3原子的缺電性增加,而N原子電子云密度增加,其原子電荷QN從-0.318 e變為-0.524 e,說明摻雜使電子從芳環轉移到鏈上N原子.由于在摻雜位形成氫鍵,存在電子在摻雜物和底物之間的轉移,造成聚苯胺環內電子的重新分布,電子密度的轉移是產生幾何構型變化的根本原因.原子電荷的計算結果很好的驗證了幾何計算結果,由于電子從環內轉向鏈間,使鏈間C-N-C的鍵角增加,相鄰環之間的兩面角減小,電子的共軛相應增強,從而提高了聚苯胺的導電性能.從N—H鍵長的計算結果可以看出,摻雜點形成的氫鍵長度為0.1105 nm,大于苯環中的氫鍵長度0.102 nm,說明摻雜形成的氫鍵強度較弱,易于通過去質子化實現去摻雜.
3.3 對甲苯磺酸摻雜態聚苯胺
按照P-TSA結構式繪制了P-TSA分子,在對分子進行結構優化的基礎上,將P-TSA分子中磺酸基中的H原子置于苯胺醌環兩側的N原子上,構建了P-TSA摻雜PANI模型,如圖5所示.為了減小摻雜大分子對苯胺結構產生空間位阻效應,建模時盡量使P-TSA分子中的苯環遠離聚苯胺分子鏈.為了同HCl摻雜作對比,摻雜方式同樣采用雙極化子摻雜.
3.3.1 幾何結構
P-TSA摻雜對聚苯胺中各鍵鍵長影響的規律同HCl摻雜一致,表現為醌環中的C=C雙鍵鍵長明顯增加,同時連接各環的C=N雙鍵鍵長亦明顯增加,說明摻雜后聚合物鏈原本具有的苯-醌交替的結構不復存在,更多地體現出一種芳香環的共軛結構.
由于摻雜對非摻雜點芳環幾何結構的影響很小,表2僅列出了各摻雜位置上C-N-C鍵角和相鄰環扭轉角的平均值計算結果.摻雜使C-N-C鍵角增大,同時使相鄰環間的扭轉角減小.對比具體的數值,可發現P-TSA摻雜方式下C-N-C鍵角的增加略大于HCl摻雜,共面性也略好于鹽酸,因此推測P-TSA摻雜比HCl摻雜具有更好的導電特性.
3.3.2 電子結構
P-TSA摻雜前后體系內各原子的原子電荷計算結果如表3所示,表中的原子標號見圖5.表中列出的數據為8個摻雜點計算結果的平均值.
P-TSA分子摻雜后,原分子中磺酸根與H原子形成的氫鍵斷裂,H原子同苯胺低聚物中的亞胺基氮結合形成氫鍵.從與P-TSA分子相關的原子電荷變化可以看出,磺酸中的H原子與亞胺基N原子形成氫鍵后,H的原子電荷從0.435e增加至0.458 e,說明摻雜后形成的氫鍵中,H的正離子型更強.磺酸分子中S原子電荷從0.637e減小至0.625 e,而與其成鍵的三個O原子電荷均有所增加,說明失去氫質子后磺酸根中S—O鍵的電子密度向O原子一邊偏移.重點考察摻雜點上的N原子,發現N的原子電荷由-0.318 e變化至-0.593 e,即摻雜使N原子的電子云密度增加0.275 e,而與氮相鄰的C2、C3原子則失去電子,說明N原子得到的電子是由苯環中的π電子提供的.與HCl摻雜后N原子電荷對比(變化量為0.206 e),可看出有機磺酸摻雜更利于電子從環內向鏈間轉移.原子電荷的計算結果同幾何優化結果一致,均從理論上說明PANI采用P-TSA摻雜可比HCl摻雜獲得更好的導電特性.
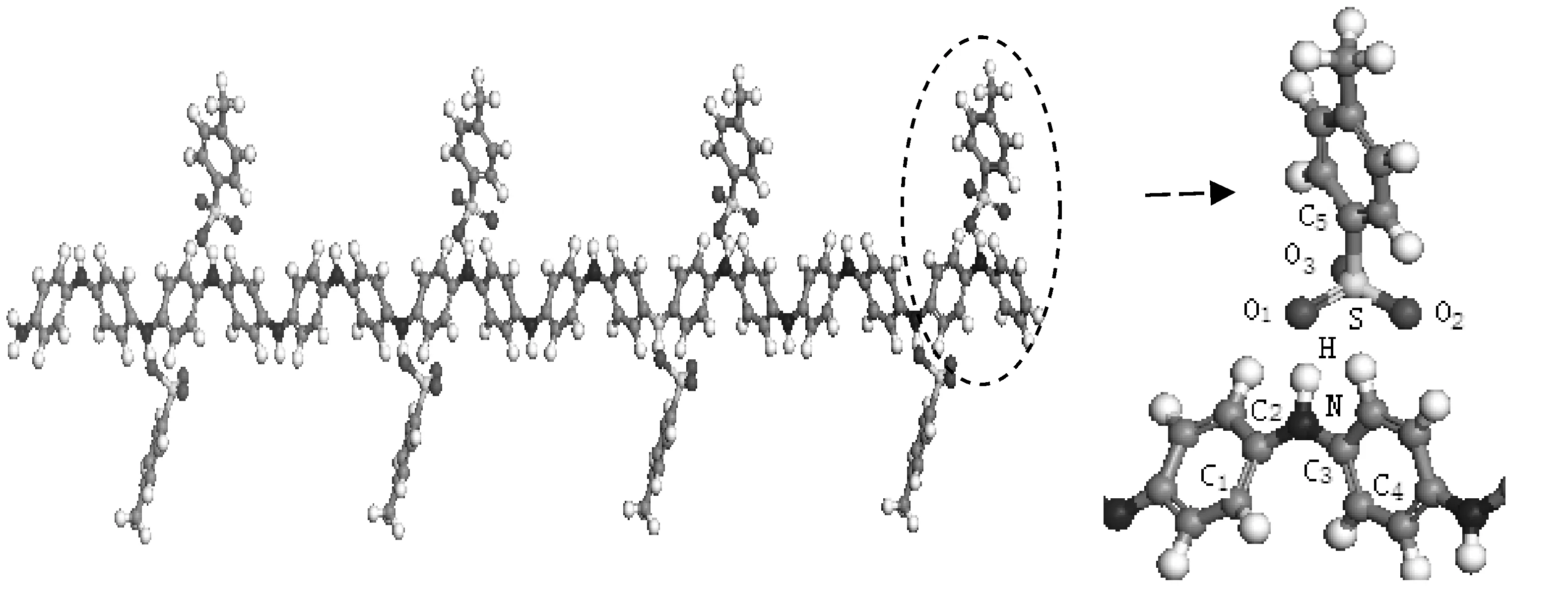
圖5 P-TSA摻雜苯胺低聚物模型Fig.5 Model of P-TSA doped Oligoaniline
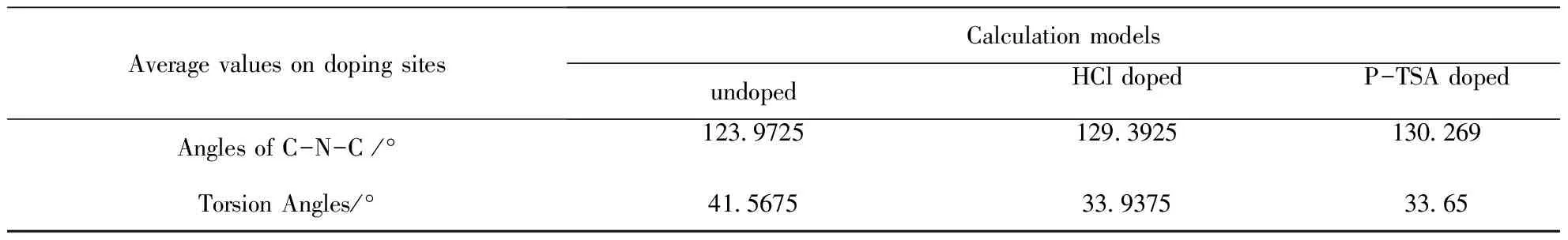
表2 P-TSA摻雜位上C-N-C鍵角和扭轉角的計算結果
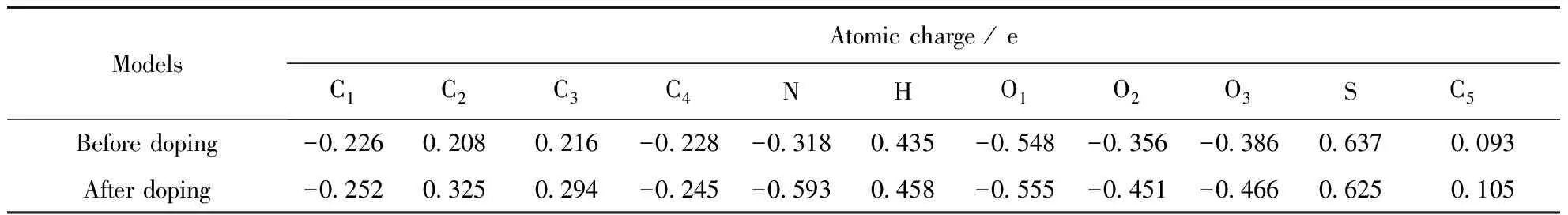
表3 P-TSA摻雜點附近的原子電荷
4 結 論
采用密度泛函理論方法,對苯胺低聚物及質子酸摻雜苯胺低聚物的結構進行了計算與分析.結果表明:(1)HCl摻雜很大程度上改變了醌環中的C-C鍵長,C-C單、雙鍵交替的性質被削弱.摻雜位置上C=N雙鍵平均鍵長由0.1323 nm增加至0.1357 nm,鏈間C-N-C鍵角平均值由123.97°增至129.39°,扭轉角平均值由41.57°減小到33.94°.摻雜位N原子電荷減少,而與其成鍵的兩個C原子電荷均有所增加,電子從芳環轉移到鏈上.(2)P-TSA摻雜改變苯胺低聚物幾何結構的規律同HCl摻雜一致,摻雜點C-N-C鍵角平均值略大于HCl摻雜,相鄰環間的共面特性略優于HCl摻雜.P-TSA摻雜點上N原子電荷變化0.275 e,大于HCl摻雜的0.206 e,理論上表明有機磺酸摻雜更利于電子從環內向鏈間轉移.
[1] Rajesh Tarushee A, Devendra K. Recent progress in the development of nano-structured conducting polymers /nanocomposites for sensor applications [J].SensorsandActuatorsB, 2009, 136: 275.[2] Masanobu M, Takuya A. Properties and stability of polyaniline nanofiber ammonia sensors fabricated by novel on-substrate method [J].SensorsandActuatorsB, 2011, 160: 999.
[3] Tai H L, Jiang Y D, Xie G Z,etal. Preparation, characterization and comparative NH3-sensing characteristic studies of PANI/inorganic Oxides nanocomposite thin films[J].JournalofMaterialsSciencesandTechnology, 2010, 7: 605.
[4] Liu H Y, Wang S B, Zhu Y,etal. Hydrothermal synthesis of polyaniline-iron oxide nano-composite materials [J].ChemicalJournalofChineseUniversities, 2013, 34(5): 1303 (in Chinese) [劉紅纓, 王雙寶, 朱英, 等. 水熱法制備聚苯胺及其鐵氧化物納米復合材料[J]. 高等學校化學學報, 2013, 34(5): 1303]
[5] Lim S L, Tana K L. A comparative ab initio and DFT study of neutral aniline oligomers [J].JournalofChemicalPhysics, 2000, 122(23): 10648.
[6] Cavazzoni C, Colle R, Farchioni R,etal. Car-Parrinello molecular dynamics study of electronic and structural properties of neutral polyanilines [J].PhysicalReviewB, 2002, 66: 165110.
[7] Foreman J P, Monkman A P. Theoretical investigations into the structural and electronic influences on the hydrogen bonding in doped polyaniline [J].TheJournalofPhysicalChemistryA, 2003, 107: 7604.
[8] Varela A, lvarez A, Sordo J,etal. Doping of polyaniline by acid-base chemistry: Density functional calculations with periodic boundary conditions [J].TheJournaloftheAmericanChemicalSociety, 2005, 127:11318.
[9] Casanovas J, Aleman C. Comparative theoretical study of heterocyclic conducting oligomers neutral and oxidized forms [J].TheJournalofPhysicalChemistryC, 2007, 111: 4823.
[10] Zhekova H, Tadjer A, Ivanova A,etal. Theoretical study of the structure and electronic spectra of fully protonated emeraldine Oligomers [J].InternationalJournalofQuantumChemistry, 2007, 107: 1688.
[11] Colle R, Parruccini P, Benassi A,etal. Optical properties of emeraldine salt polymers from ab initio calculations: Comparison with recent experimental data. [J].TheJournalofPhysicalChemistryB, 2007, 111: 2800.
[12] Cavazzoni C, Colle R, Farchioni R,etal. Acidification of three-dimensional emeraldine polymers: Search for minimum energy paths from base to salt [J].JournalofChemicalPhysics, 2008, 128: 234903.
[13] Aleman C, Ferreira C A, Torras J,etal. On the molecular properties of polyaniline: A comprehensive theoretical study [J].Polymer, 2008, 49: 5169.
[14] Yang G, Hou W, Feng X O,etal. Electronic structure of oligoaniline doped by inorganic and organic acids [J].InternationalJournalofQuantumChemistry, 2008, 108: 1155.
[15] Casanovas J, Canales M, Ferreira C A,etal. A first principle analysis of the structure of oligoanilines doped with alkylsulfonic acids [J].TheJournalofPhysicalChemistryA, 2009, 113: 8795.
[16] Zhang H F, Zhang S D, Kong X H. Vibrational spectrum of C6H5NH2in S1state and ab initio calculations[J].JournalofAtomicandMolecularPhysics(原子與分子物理學報), 2010, 27(4): 643.
[17] Paterno L G, Mattoso L H C. Influence of different dopants on the ddsorption, morphology, and properties of self-assembled films of Poly(o-ethoxyaniline) [J].JournalofAppliedPolymerScience, 2002, 83: 1309.
[18] MacDiarmid A G, Chiang J C, Richter A F,etal. Polyanilne: a new concept in conducting polymers [J].SyntheticMetals, 1987, 18(1-3): 285.
[19] MacDiarmid A G, Epstein A J. Secondary doping in polyanilne[J].SyntheticMetals, 1995, 69: 22.
[20] Accelrys Incorporation, Material Studio, Accelrys Inc: SanDiego, CA.
[21] Zhang F L, Wu X F. Density functional theory study of 3-octylthien-2, 5-ylenediethynylene-co- benzo[c]- 1’2’ 5’- thiadiazo-3, 6-ylenedi adsorption on TiO2(100) Surface[J].JournalofAtomicandMolecularPhysics, 2012, 29(5): 927 (in Chinese) [張福蘭, 吳興發. 4, 7-二(2-噻吩基)苯并噻二唑-3-辛基噻吩二炔在TiO2(100)表面吸附的密度泛函理論研究[J].原子與分子物理學報, 2012, 29(5): 927]
Density functional theory study of the structures of oligoaniline and doped-oligoaniline
XUE Yan-Bing1, YU Jing-Yi1, XU Zhi1, TANG Zhen-An2
(1. School of Electrical and Information, Dalian JiaoTong University, Dalian 116028, China 2.School of Electronic Science and Technology, Dalian University of Technology, Dalian 116024, China)
A comprehensive study about the geometric structure and electronic structure of aniline oligomer and hydrochloric acid, p-toluene sulfonic acid doped oligomer has been developed using theoretical calculation method based on density functional theory. The results show that the property of C-C bond length of quinone ring in Oligoaniline molecular chains alternating in the way of single and double bond is obviously weakened by doped proton-acid, and the bond length of C=N between chains is significantly increased. The bond angle of C-N-C on doping site is increased and the torsion angle between adjacent rings is reduced. Coplanar of the molecular is improved by acid doping. Compared with HCl, doping with p-toluene sulfonic acid is more conducive to transfer electron from the aromatic rings to the molecular chains, so doping with organic acid can better improve the conductivity of Polyaniline material theoretically.
Oligoaniline; Density functional method; Geometrical structure; Electronic structure
103969/j.issn.1000-0364.2015.02.005
2013-11-25
國家自然科學基金(61201092);遼寧省自然科學基金(201202015);遼寧省高等學校優秀人才支持計劃(LJQ2013047)
薛嚴冰,女,博士,教授,碩士生導師,主要從事半導體氣體傳感器方面的研究. E-mail: dlxyb@djtu.edu.cn
O649.5
A
1000-0364(2015)02-0201-06
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
