3種黃酮醇ESI-ITMS準分子離子構型的密度泛函理論分析
2015-04-27 03:12:06孫長海姜文婷房碧晗王慧冰方洪壯
分析測試學報 2015年2期
李 想,孫長海,姜文婷,房碧晗,王慧冰,方洪壯
(佳木斯大學 藥學院,黑龍江 佳木斯 154007)
3種黃酮醇ESI-ITMS準分子離子構型的密度泛函理論分析
李 想,孫長海,姜文婷,房碧晗,王慧冰,方洪壯*
(佳木斯大學 藥學院,黑龍江 佳木斯 154007)
采用密度泛函理論(Density functional theory,DFT) B3LYP方法,通過全幾何結構優化、二面角柔性勢能掃描,對山奈酚、槲皮素、楊梅素3種黃酮醇分子的優勢構象及電噴霧離子阱質譜(ESI-ITMS)負離子模式下準分子離子的最優構型進行了研究,并從能量參數、構型參數、質譜實驗角度對準分子離子的最優構型作出了確證。結果表明:山奈酚、槲皮素和楊梅素分子二面角D(1,2,1′,6′)均接近0°,分子的優勢構象均為B環與A,C環處在同一平面上的構象;在負離子模式下,自動及手動掃描方式得到的山奈酚、槲皮素、楊梅素的二級質譜的復雜程度依次減弱,各分子失去羥基氫所形成的準分子離子結構共軛鏈增長、共軛效應加強;山奈酚存在兩種低能量的準分子離子構型,即失去B環4′位羥基氫與失去A環7位羥基氫的構型,槲皮素與楊梅素失去B環4′位羥基氫的總能量最低、構型最穩定,且山奈酚、槲皮素、楊梅素準分子離子構型的穩定性依次增加。該研究可供進一步探索黃酮醇類化合物ESI-ITMS負離子模式下的質譜裂解規律參考。
黃酮醇化合物;電噴霧離子阱質譜;密度泛函理論
黃酮醇(Flavonol)屬黃酮類化合物,廣泛分布于自然界,常以游離態或糖苷形式存在[1-3],具有抗突變、抗腫瘤、抗氧化、延緩衰老等多種藥理和保健作用,被廣泛用于藥品和保健食品的開發[4-6]。黃酮醇類化合物的天然產物及體內樣品的分析[7-8]方法多采用電噴霧電離源質譜技術。隨著軟電離質譜的不斷發展,借助量子化學方法從分子水平研究化合物軟電離方式下的質譜裂解規律受到廣泛關注[9-10]。在質譜裂解規律的量子化學計算中,準分子離子幾何結構的可靠性直接影響后續深層次的分析,確定準分子離子最有可能的最優構型是解析質譜裂解機理的首要問題[11-12]。本研究運用量子化學計算方法,依據密度泛函理論(Density functional theory,DFT),借助Gaussian 03計算軟件,考察了山奈酚、槲皮素、楊梅素3種黃酮醇分子處于穩定狀態時的結構特征,探索了各化合物分子的優勢構象以及電噴霧電離源負離子模式下準分子離子的最優構型,結合構型參數與質譜測定,對獲得的準分子離子最優構型進行了確證。實驗結果可供深入探索ESI-ITMS負離子模式下黃酮醇類化合物的質譜裂解機理參考。
1 實驗部分
1.1 儀器與試劑
AgilentG6310電噴霧離子阱質譜儀(美國安捷倫公司);KDS 100 CE微量注射泵(Agilent kd scientific)。對照品:山奈酚(Kaempferol,純度>98%,批號20121215,大連美侖生物技術有限公司);槲皮素(Quercetin,純度>98%,批號1109630,天津一方科技有限公司);楊梅素(Myricetin,純度≥98%,批號13020102,成都曼斯特生物科技有限公司)。甲醇、異丙醇(色譜級,天津市科密歐化學試劑有限公司)。
1.2 質譜條件
離子源:ESI源;掃描方式:負離子模式下自動一級質譜全掃描及手動二級質譜提取離子掃描;碎裂電壓:自動1.00 V,手動0.10~1.50 V;掃描范圍:m/z50~330;鞘氣和輔助氣:氮氣;鞘氣流速10 L·min-1;輔助氣流速:0.15 L·min-1;碰撞氣:氦氣;噴霧電壓:3.5 kV;毛細管電壓:13 V;干燥氣溫度:325 ℃;微量注射泵進樣流速:0.5 mL·h-1;進樣方式:直接進樣。對照品濃度:5 μg/mL;溶劑:甲醇。
1.3 量子化學計算
量子化學計算采用Gaussian 03軟件完成。各化合物分子及離子的初始幾何結構由軟件GaussView 3.0給出。首先,采用密度泛函理論B3LYP/6-31+g(2d,2p)方法,對分子初始結構中B環與C環間的二面角進行旋轉360°柔性勢能掃描,獲得各分子能量最低的構象;再用B3LYP/6-31g(d)方法對獲得的構象進行優化,得到各分子的優勢構象及相關參數。在優勢構象基礎上,采用相同優化方法對各分子可能的準分子離子構型進行結構優化及振動分析,并在RB3LYP/6-31+g(2d,2p)水平計算各個優化結構的能量;最后,通過能量比較分析,得出各化合物準分子離子的最優構型。結構優化及振動分析收斂標準均為:力的最大值(Maximum Force)小于0.000 450;力的均方根(RMS Force)小于0.000 300;位移的最大值(Maximum Displacement)小于0.001 800;位移的均方根(RMS Displacement)小于0.001 200。鍵長、偶極矩等參數從各分子及準分子離子相應的優化結果文件中獲取。
2 結果與討論
2.1 分子優勢構象
黃酮醇化合物分子的初始結構及原子位次編號如圖1所示,其中,山奈酚:R1=H,R2=H;槲皮素:R1=OH,R2=H;楊梅素:R1=OH,R2=OH。分子柔性勢能掃描各角度構象與最穩定構象的能量差結果如圖2所示。由圖2可知,當分子處于最穩定狀態即能量最低時,山奈酚、槲皮素和楊梅素的二面角D(1,2,1′,6′)均接近0°。各分子能量最低結構的優化結果均符合收斂標準,振動分析未出現虛頻,各構象穩定。各分子的優勢構象如圖3所示。3種化合物優勢構象的部分二面角參數見表1。表1數據表明,當分子構型相對最穩定時,A,C,B環處于同一平面,與文獻[13-14]報道相吻合。

圖1 3種化合物的分子結構
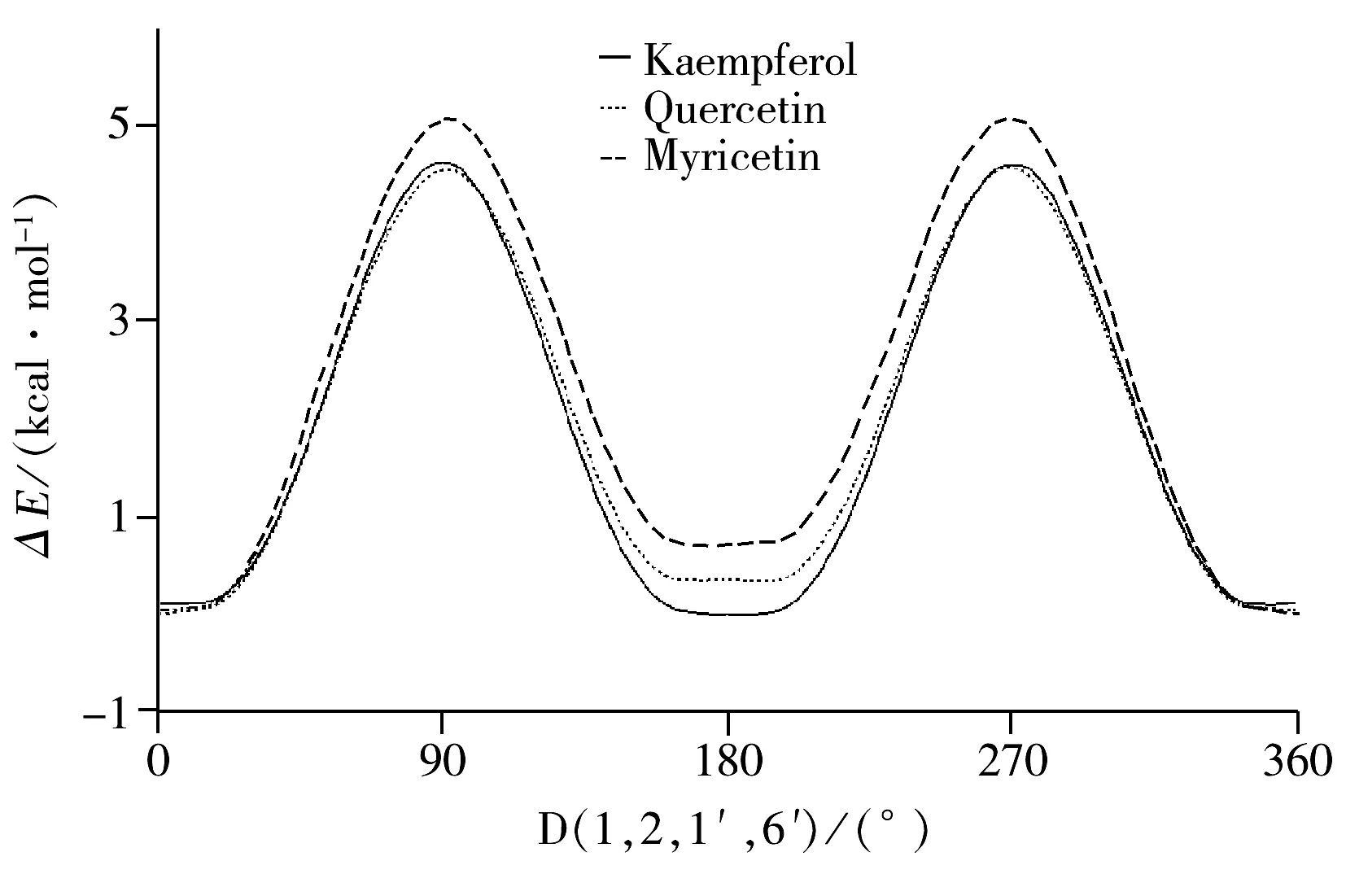
圖2 3種化合物二面角D(1,2,1′,6′)的柔性勢能掃描曲線

圖3 3種化合物的優勢構象
表1 3種化合物優勢構象的部分二面角參數
Table 1 Some dihedral parameters of the preferred conformations of three compounds
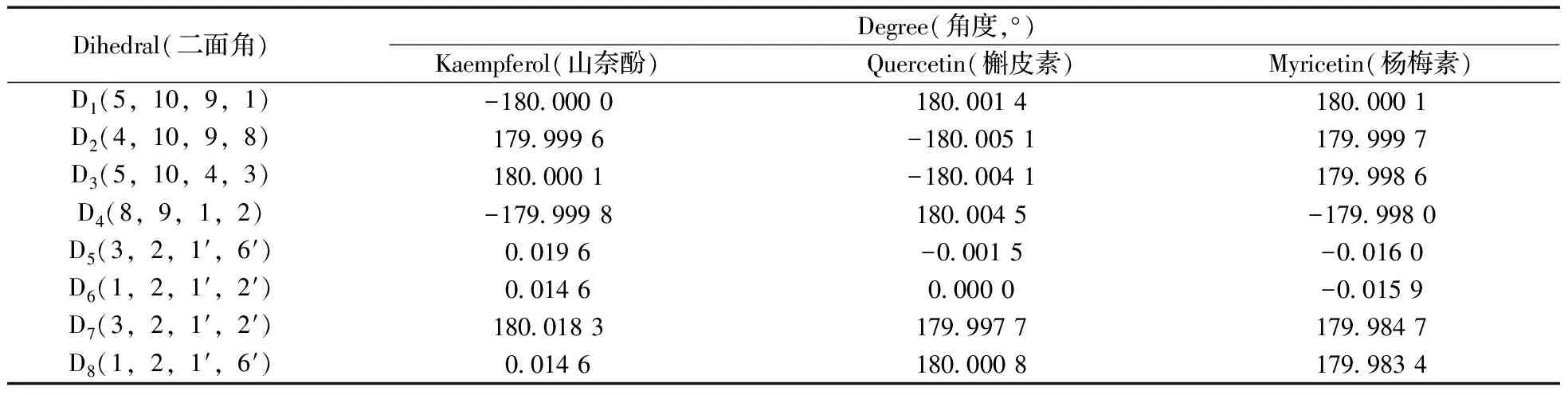
Dihedral(二面角)Degree(角度,°)Kaempferol(山奈酚)Quercetin(槲皮素)Myricetin(楊梅素)D1(5,10,9,1)-180 0000180 0014180 0001D2(4,10,9,8)179 9996-180 0051179 9997D3(5,10,4,3)180 0001-180 0041179 9986D4(8,9,1,2)-179 9998180 0045-179 9980D5(3,2,1′,6′)0 0196-0 0015-0 0160D6(1,2,1′,2′)0 01460 0000-0 0159D7(3,2,1′,2′)180 0183179 9977179 9847D8(1,2,1′,6′)0 0146180 0008179 9834
分子初始結構的輸入方法會影響優勢構象的結果。當采用ChemDraw Ultra8.0及Chem3D Ultra8.0方法時,優化后山奈酚、槲皮素和楊梅素的分子B環與A,C環的二面角分別為36.5°,36.1°和36.4°;而通過GaussView3.0輸入,對二面角D(1,2,1′,6′)進行柔性勢能掃描,得到各分子優勢構象所對應的B環與A,C環的二面角卻均接近0°。由于柔性勢能掃描能夠獲得D(1,2,1′,6′)旋轉1周的各個角度的能量值,可通過能量的比較來確定分子的優勢構象,相對更加全面合理,因此本研究采用GaussView3.0輸入。需指出,雖然兩種輸入方式產生的優勢構象結果不同,但相應的優勢構象間的能量差別較小,在室溫及空間位阻很小的情況下,非平面構象只需克服很小的能壘即可依靠分子熱運動所提供的能量轉化成平面構象[15]。
2.2 準分子離子結構
2.2.1 能量分析 在電噴霧電離源負離子模式下,羥基氫較苯環氫更易丟失而發生去質子化。山奈酚、槲皮素、楊梅素分子中分別具有4,5,6個羥基,可分別失去質子而形成準分子離子([M-H]-)。3種化合物可能的[M-H]-構型的結構優化、振動分析及能量計算表明:優化均符合收斂標準,振動分析無虛頻,構型穩定。表2為3種黃酮醇可能的[M-H]-體系的能量計算結果,可知山奈酚、槲皮素與楊梅素丟失B環04′-H(即4′位羥基氫,下同)的體系總能量均最低,構型最穩定,所形成[M-H]-為熱力學擇優結果。
與此同時,由于山奈酚丟失A環07-H(即7位羥基氫,下同)與丟失B環04′-H所形成的兩種[M-H]-總能量接近,故在負離子模式下,山奈酚存在兩種不同的[M-H]-構型,分別記作山奈酚Ⅰ(Kaempferol Ⅰ,丟失04′-H)與山奈酚Ⅱ(Kaempferol Ⅱ,丟失07-H)。
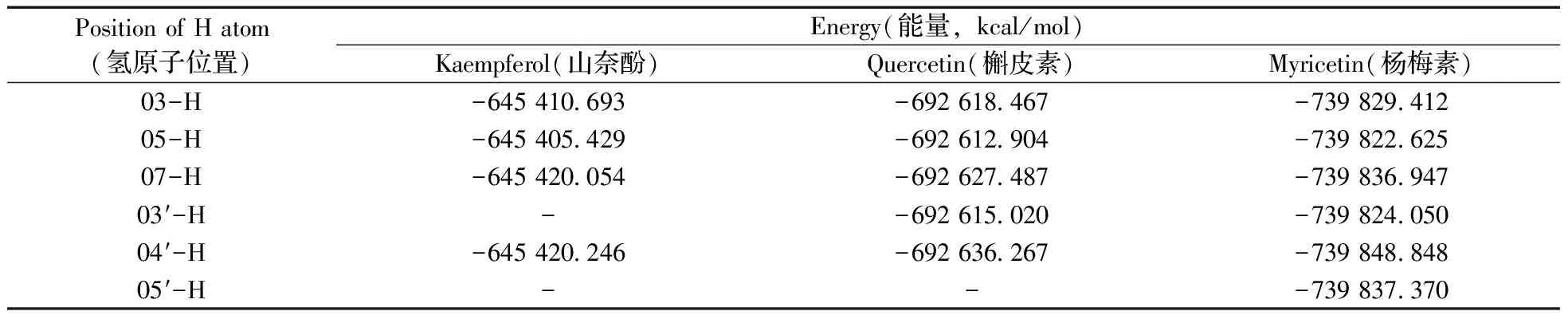
表2 3種化合物可能的[M-H]-構型的能量
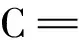
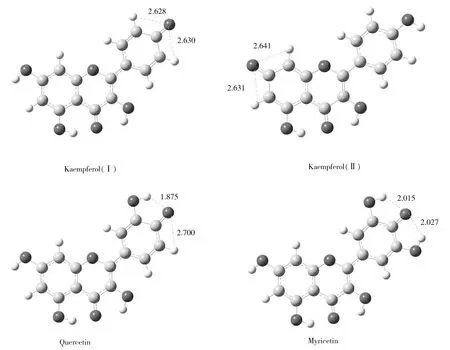
圖4 3種化合物丟失04′-H的[M-H]-立體構型及可能的氫鍵(?)
2.2.3 質譜分析 3種黃酮醇的自動二級質譜圖如圖5所示。由圖5可知,山奈酚、槲皮素、楊梅素二級質譜的復雜程度逐漸降低。對各準分子離子施以不同碎裂電壓,手動提取二級質譜時發現,當碎裂電壓較低時,山奈酚即可獲得多個產物離子,槲皮素只產生兩個明顯的離子(m/z178.8與m/z150.8),而楊梅素僅有一個明顯的m/z178.8離子。由于二級質譜所呈現的復雜程度是隨著化合物準分子離子穩定性的升高而降低,因此可以說明山奈酚、槲皮素、楊梅素準分子離子的穩定性逐漸增加。
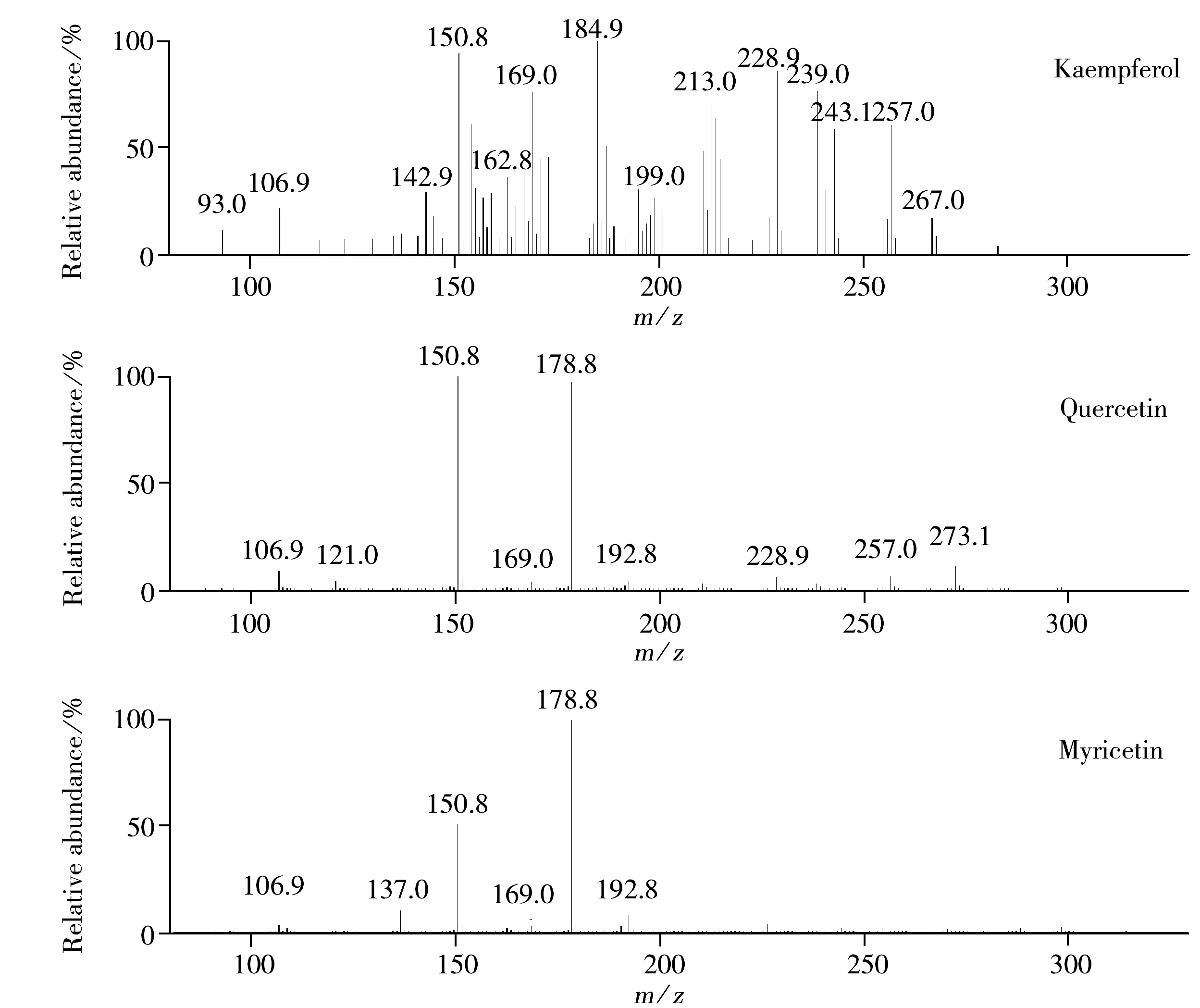
圖5 3種化合物[M-H]- 的MS2質譜圖
3種黃酮醇的[M-H]-穩定構型的相應偶極矩計算結果為:山奈酚(Ⅰ)11.697 4D、山奈酚(Ⅱ)13.230 9D、槲皮素10.058 1D、楊梅素9.022 5D。結合質譜測定結果及相關文獻資料分析可推斷出,對于[M-H]-最為穩定的楊梅素,由于分子C環極性相對較小,使得二級質譜主要發生C環跨環開裂產生1,2A-m/z178.8離子,進而脫去中性分子CO生成m/z150.8離子及連續脫去CO 和CO2后的m/z106.9離子[16]。槲皮素[M-H]-的極性相對于楊梅素有所下降,在C環跨環開裂產生m/z178.8,150.8,106.9離子的同時,也發生C環的另一跨環開裂,產生1,3A-m/z150.8離子[16-17],使得m/z150.8處的強度增大;與此同時,C環同環開裂的程度增加,有多個脫去中性低質量碎片后的離子產生。山奈酚[M-H]-極性相對較大,其穩定性較差,故其二級質譜無1,2A-m/z178.8離子產生,而在產生1,3A-m/z150.8離子的同時,還產生另一跨環開裂的0,2A-m/z162.8離子及其連續碎裂后的m/z106.9離子,并且C環同環開裂的比例加大,產生大量脫去中性碎片的離子[18-20]。
3種黃酮醇[M-H]-體系的C環各鍵鍵長及C環與B環相接的鍵長見表3。由表3可知,構成C環的C—C鍵與C—O鍵及連接C環與B環的C—C鍵的長度介于單鍵與雙鍵之間,說明組成C環的各原子間及C環與B環間存在著共軛現象。由表3還知,在3種黃酮醇[M-H]-所涉及的化學鍵中,山奈酚、槲皮素、楊梅素多數鍵鍵長的順序是由長到短。由于鍵長越長,鍵越易斷裂,從而可進一步解釋3種黃酮醇質譜復雜的順序。結合3種黃酮醇的二級質譜可知,由于C環的各鍵中,C2—C3及C4—C10鍵的鍵長數值最大,其中又以山奈酚為最大,因此山奈酚[M-H]-最易發生C2—C3及C4—C10斷裂,在質譜中表現為,先脫去中性分子CO生成m/z257.0離子,進而再分別脫去H2O與CO生成m/z239.0與m/z228.9的離子,m/z228.9離子可繼續脫去CO2生成m/z184.9的離子。另外,山奈酚C4—C10的斷裂,也可導致C2H2O的脫去,產生m/z243.1離子,進而脫去CO2生成m/z199.0的離子;同時C4—C10的斷裂還會引發m/z142.9離子的產生。由表3還可知,3種黃酮醇[M-H]-連接C環與B環的C2—C1′鍵較長、較易斷裂,表現在圖5的二級質譜上,山奈酚脫去中性碎片AC環,產生m/z93.0的酚羥基離子[21],而槲皮素和楊梅素[M-H]-脫去中性碎片B環,產生m/z192.8的離子[16]。雖然m/z192.8的離子峰強度較低,但此峰的強度與槲皮素和楊梅素[M-H]-的鍵長順序相對應。關于山奈酚與另外兩種黃酮醇在C2—C1′鍵開裂時生成不同產物離子的現象,推測其原因為,山奈酚[M-H]-構型Ⅱ的C2—C1′鍵鍵長大于構型Ⅰ,鍵斷裂發生在構型Ⅱ上,鍵斷裂時電荷轉移的機制與丟失04′-H構型的槲皮素和楊梅素的[M-H]-有所不同。

表3 3種化合物[M-H]-C環的主要鍵長(?)
3 結 論
依據密度泛函理論,通過量子化學計算確定山奈酚、槲皮素、楊梅素化合物分子的優勢構象為A,C,B環處在同一平面,為平面共軛體系。量子化學與質譜實驗的結合分析表明,電噴霧離子阱質譜負離子模式下,3種黃酮醇分子中B環4′-OH脫去質子及山奈酚A環7-OH脫去質子所形成的準分子離子體系,使共軛系統共軛鏈增長,共軛效應加強,總能量最低,均為準分子離子的最優構型。
[1] Day A J,Bao Y,Morgan M R,Williamson G.FreeRadicalBiol.Med.,2000,29(12):1234-1243.
[2] Ablajan K,Abliz Z,Shang X Y,He J M,Zhang R P,Shi J G.J.MassSpectrom.,2006,41(3):352-360.
[3] Manach C,Morand C,Demigné C,Texier O,Régérat F,Rémésy C.FEBSLett.,1997,409(1):12-16.
[4] Vuorinen H,M??tt? K,T?rr?nen R.J.Agric.FoodChem.,2000,48(7):2675-2680.
[5] Garcia-Closas R,Gonzalez C A,Agudo A,Riboli E.CancerCauses&Control,1999,10(1):71-75.
[6] Su Y,Liu S,Yang M,Liao X W.J.SouthwestUniv.Natl.:Nat.Sci.Ed.(蘇宇,劉珊,楊銘,廖顯威.西南民族大學學報:自然科學版),2006,32(3):517-521.
[7] Geng P,Zhang R,Aisa H A,He J,Qu K,Zhu H,Abliz Z.RapidCommun.MassSpectrom.,2007,21(12):1877-1888.
[8] Chi Y M,Zhu H Y,Ju L,Zhang Y,Shen X N,Hua X Y,Nie F.Chin.J.Anal.Chem.(池玉梅,朱華云,居羚,張瑜,沈小寧,華小懿,聶芬.分析化學),2009,37(2):227-231.
[9] Wright P,Alex A,Harvey S,Parsons T,Pullen F.Analyst,2013,138(22):6869-6880.
[10] Galezowska A,Harrison M W,Herniman J M,Skylaris C K,Langley G J.RapidCommun.MassSpectrom.,2013,27(9):964-970.
[11] Ouyang Y Z,Liang Y Z,Li S H,Lou X,Zhang L X,Tang Z H,Wang Q,Xu X N.Int.J.MassSpectrom.,2009,286(2/3):112-121.
[12] Risoli A,Cheng J B,Verkerk U H,Zhao J,Ragno G,Hopkinson A C,Siu K W.RapidCommun.MassSpectrom.,2007,21(14):2273-2281.
[13] Aparicio S.Int.J.Mol.Sci.,2010,11(5):2017-2038.
[14] Antonczak S.J.Mol.Struct.Theochem.,2008,856(1/3):38-45.
[15] Cornard J P,Merlin J C,Boudet A C,Vrielynck L.Biospectroscopy,1997,3(3):183-193.
[16] Fabre N,Rustan I,Hoffmann E,Quetin-Leclercq J.J.Am.Soc.MassSpectrom.,2001,12(6):707-715.
[17] Burns D C,Ellis D A,Li H X,Lewars E,March R E.RapidCommun.MassSpectrom.,2007,21(3):437-454.
[18] Medana C,Carbone F,Aigotti R,Appendino G,Baiocchi C.Phytochem.Anal.,2008,19(1):32-39.
[19] Lu L,Song F R,Tsao R,Jin Y R,Liu Z Q,Liu S Y.RapidCommun.MassSpectrom.,2010,24(1):169-172.
[20] March R E,Miao X S.Int.J.MassSpectrom.,2004,231(2/3):157-167.
[21] Hughes R J,Croley T R,Metcalfe C D,March R E.Int.J.MassSpectrom.,2001,210/211:371-385.
Analysis on ESI-ITMS Quasi-molecular Ion Configurations of Three Flavonol Compounds by Density Functional Theory
LI Xiang,SUN Chang-hai,JIANG Wen-ting,FANG Bi-han,WANG Hui-bing,FANG Hong-zhuang*
(Pharmaceutical College,Jiamusi University,Jiamusi 154007,China)
Using density functional theory(DFT) B3LYP method,the preferred molecular conformations and the optimal configurations of kaempferol,quercetin and myricetin quasi-molecular ions in electrospray ion trap mass spectrometry(ESI-ITMS) negative ion mode were studied by the full geometry optimization and the relaxed dihedral potential energy scan.The optimal configurations of three flavonol quasi-molecular ions were confirmed in the aspects of energy parameters,configuration parameters,and mass spectrometry experiments.The results indicated that the degrees of dihedral D(1,2,1′,6′) for kaempferol,quercetin and myricetin were close to 0°,and the preferred conformations of three flavonol compounds were respectively coplanar including A,C and B ring.In the negative ion mode,the complexities of MS2spectra which were determined by the automatic and manual methods for kaempferol,quercetin and myricetin were successively weakened.The optimal configurations of quasi-molecular ions were the structures lost the hydrogen of phenolic hydroxyl,with the conjugated chain increasing and the conjugated effect strengthening.Two quasi-molecular ion configurations for kaempferol existed,that were loss of 4′-OH hydrogen and 7-OH hydrogen,and the total energy of loss of 4′-OH hydrogen configurations for quercetin and myricetin were the lowest and the most stable.The stabilities of quasi-molecular ion configurations for kaempferol,quercetin and myricetin were increased successively.The analysis of the study can be used as a reference for further exploration of the fragmentation pathway and mechanism of flavonol compounds in ESI-ITMS negative ion mode.
flavonol compounds; electrospray ionization ion trap mass spectrometry(ESI-MS); density functional theory
2014-10-11;
2014-11-10
佳木斯大學研究生科技創新項目(LZR2014_032);黑龍江省自然科學基金資助項目(D201226)
10.3969/j.issn.1004-4957.2015.02.004
O657.63;TQ460.72
A
1004-4957(2015)02-0147-06
*通訊作者:方洪壯,教授,研究方向:中藥分析與計算藥物分析,Tel:0454-8611265,E-mail:fhz-chjms@sohu.com
