黃麥顆粒的質量標準研究Δ
2015-05-21 08:55:52金冠欽夏玲紅孫黎林厚文上海交通大學醫學院附屬仁濟醫院上海20000上海交通大學醫學院上海200025
中國藥房 2015年24期
金冠欽,夏玲紅,孫黎#,林厚文,2(.上海交通大學醫學院附屬仁濟醫院,上海 20000;2.上海交通大學醫學院,上海 200025)
黃麥合劑是上海交通大學醫學院附屬仁濟醫院院內制劑(滬藥制字Z05060356),由黃芪、淫羊藿、制何首烏、菟絲子、麥冬等7味中藥材提取制成[1],具有滋陰益氣、溫腎壯陽的功效,用于男性少、弱精癥的治療[2-6]。其中,菟絲子、淫羊藿為君藥,黃芪、何首烏為臣藥。本品使用30余載,療效確切[7],廣受病患及醫護人員的好評。由于黃麥合劑的制劑類型存在不易于儲存和攜帶、有效成分含量較低的缺點,故我院依托上海市科學技術委員會科研計劃科技支撐項目,對黃麥合劑進行劑型改造,從合劑改為更加易于攜帶的顆粒劑,并在此過程中,對實驗生產的各批次樣品進行了質量標準研究。本試驗采用薄層色譜(TLC)法對處方中的黃芪進行定性鑒別,并以淫羊藿的主要成分淫羊藿苷、菟絲子的主要成分金絲桃苷及制何首烏的主要成分二苯乙烯苷為定量指標,研究了高效液相色譜(HPLC)法測定以上3種成分的方法,并分別對其進行方法學考察,為改進和提高黃麥顆粒的質量標準提供了方法學基礎。
1 材料
1.1 儀器
1525型HPLC儀,包括2487型紫外檢測器、breeze色譜工作站(美國Waters公司);AE 240型電子天平(瑞士Mettler-Toledo公司);RE-52型旋轉蒸發儀(上海青浦滬西儀器廠);SB5200型超聲儀(上海Branson公司);ZF-1型三用紫外分析儀(上海寶山顧村電光儀器廠);DZF-6020型真空干燥箱(上海精宏實驗設備有限公司);硅膠G薄層板(青島海洋化工廠分廠)。
1.2 藥品與試劑
黃麥顆粒(上海方心科技研究所,批號:20131218,規格:8 g/袋);黃芪甲苷對照品(批號:110781-200613,純度≥98%)、淫羊藿苷對照品(批號:121032-200501,純度≥98%)、金絲桃苷對照品(批號:111521-201004,純度≥98%)、二苯乙烯苷對照品(批號:110844-200606,純度≥99.8%)均由中國食品藥品檢定研究院提供。乙腈(色譜純,美國Merck公司,批號:1520530004);甲醇、氯仿、正丁醇、乙醚、乙酸乙酯、冰醋酸、無水乙醇均為分析純;水為蒸餾水。
2 方法與結果
2.1 黃芪的TLC鑒別
2.1.1 供試品溶液的制備 取黃麥顆粒7 g,用80ml蒸餾水超聲(功率:200 W,頻率:20 kHz)處理10min,將溶液用60ml乙醚振搖,棄去乙醚液,水層用水飽和的正丁醇液振搖提取2次,每次80ml,合并正丁醇液;用氨試液80ml洗滌,再用80ml正丁醇飽和水溶液洗滌,取正丁醇層,于100℃水浴將正丁醇液蒸干,殘渣加甲醇1ml使溶解,作為供試品溶液。
2.1.2 對照品溶液的制備 精密稱取黃芪甲苷對照品適量,加甲醇制成每1ml含0.4mg的溶液,得對照品溶液。
2.1.3 陰性對照溶液的制備 取除黃芪外的其余藥材,按黃麥顆粒處方比例及制備工藝制成缺黃芪的陰性對照品,按“2.1.1”項下方法制備成缺黃芪的陰性對照溶液。
2.1.4 結果 按TLC法[10]試驗,吸取“2.1.1”項下供試品溶液、“2.1.2”項下對照品溶液、“2.1.3”項下陰性對照溶液各10μl,分別點于同一硅膠G薄層板上,以氯仿-甲醇-水(13∶7∶2,V/V/V)于10℃以下放置分層的下層溶液為展開劑,上行展開,展距為8 cm,取出,晾干,噴以10%硫酸乙醇溶液,在105℃加熱至斑點顯色清晰,置日光下檢視。結果,供試品色譜中,在與對照品色譜相應的位置上,顯相同的粉紅色斑點;而陰性對照溶液在相應的位置上無干擾,結果見圖1。
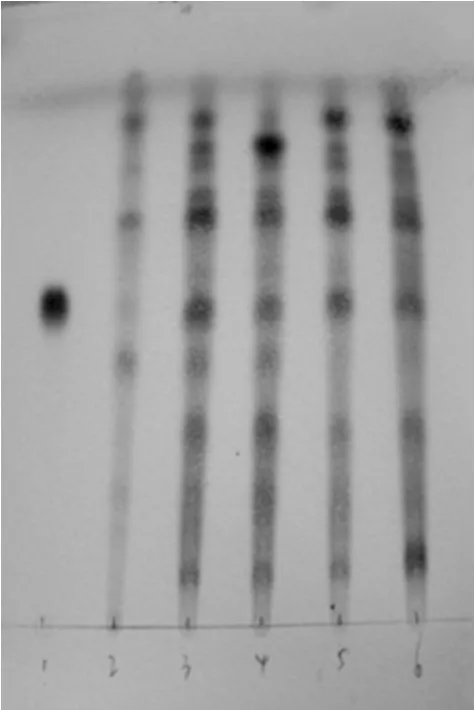
圖1 黃芪的薄層色譜圖1.黃芪甲苷對照品;2.缺黃芪的陰性對照溶液;3~6.供試品Fig 1 TLC chromatograms of Leguminosae1.reference of astragaloside A;2.negative control solution without of Leguminosae;3-6.test sample
2.2 淫羊藿苷的含量測定
2.2.1 色譜條件 色譜柱:Diamonsil-C18(250mm×4.6mm,5 μm);流動相:乙腈-水-冰醋酸溶液(30∶70∶1,V/V/V);流速:1.0ml/min;檢測波長:270nm;柱溫:30 ℃;進樣量:5μl[3]。
2.2.2 對照品溶液的制備 精密稱取淫羊藿苷對照品3.19mg,置于10ml量瓶中,用甲醇逐級稀釋成9.969、19.938、39.875、79.75、159.5、319.0μg/ml的對照品溶液。
2.2.3 供試品溶液的制備 取黃麥顆粒7.5 g,加入蒸餾水150ml攪拌均勻,70℃水浴15min,放冷,用濾紙濾過;取濾液15ml,用乙酸乙酯60、40、40ml振搖提取3次,合并乙酸乙酯層,用10ml的2.5%Na2CO3洗滌,靜置過夜。取乙酸乙酯層,80℃水浴蒸干,殘渣用甲醇定容至10ml,經0.22 μm微孔濾膜濾過,取續濾液,即得。
2.2.4 陰性對照溶液的制備 按照黃麥顆粒處方比例,制備不含淫羊藿的陰性樣品,精密稱取7.5 g,按“2.2.3”項下方法操作,即得。
2.2.5 專屬性試驗 在“2.2.1”項色譜條件下,淫羊藿苷和其他組分色譜峰線基本分離,分離度大于1.5;同時取陰性樣品進樣,結果陰性樣品溶液在淫羊藿苷色譜峰位置處無相應峰出現。色譜見圖2。
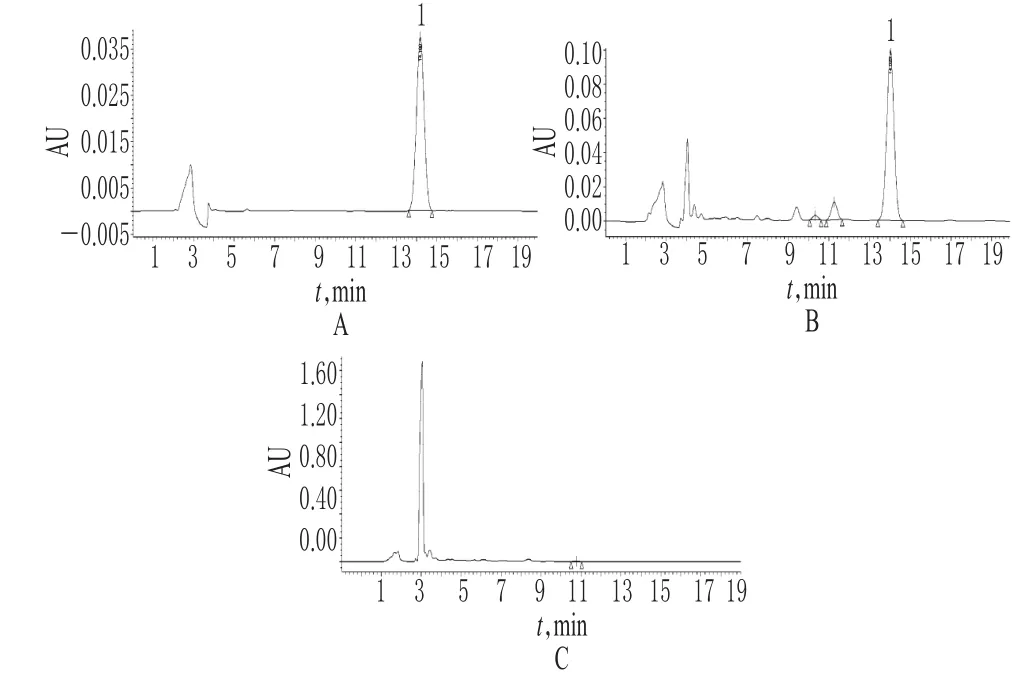
圖2 淫羊藿苷的高效液相色譜圖A.對照品(39.875μg/ml);B.供試品;C.缺淫羊藿的陰性對照;1.淫羊藿苷Fig 2 HPLC chromatograms of icariinA.control substance(39.875 μ g/ml);B.test sample;C.negative solution without Epimedium davidii;1.E.davidii
2.2.6 線性關系考察 精密量取不同質量濃度的淫羊藿苷對照品溶液9.969、19.938、39.875、79.75、159.5、319.0μg/ml各5μl,注入HPLC儀,按“2.2.1”項下色譜條件進樣測定,記錄淫羊藿苷的峰面積。以淫羊藿苷質量濃度(x,μg/ml)為橫坐標、峰面積(y)為縱坐標進行線性回歸,得回歸方程為y=2.01337×10-5x+0.896281(r=0.9999)。結果表明,淫羊藿苷質量濃度在9.969~319.0μg/ml范圍內與其峰面積積分值呈良好的線性關系。
2.2.7 精密度試驗 精密吸取“2.2.2”項下質量濃度為159.5μg/ml的對照品溶液5μl,按“2.2.1”項下色譜條件進樣測定,連續進樣6次。結果,RSD為1.51%(n=6),表明儀器精密度良好。
2.2.8 穩定性試驗 取黃麥顆粒(批號:20131218)7.5 g,按“2.2.3”項下方法制備供試品溶液,分別于配制后0、2、4、6、8 h時按“2.2.1”項下色譜條件進樣測定。結果,RSD為1.45%(n=5),表明供試品溶液在配制后8 h內穩定性良好。
2.2.9 重復性試驗 取批號為20131218的黃麥顆粒適量,按“2.2.3”項下方法配制得到6份供試品溶液,按“2.2.1”項下色譜條件進樣測定。結果,RSD為2.41%(n=6),表明本方法重復性較好。
2.2.10 加樣回收率試驗 精密稱取已知含量的黃麥顆粒樣品(批號:20131218)6份,精密加入淫羊藿苷對照品適量,按“2.2.3”項下方法制備供試品溶液,再按“2.2.1”項下色譜條件進樣測定,記錄峰面積,并計算加樣回收率,結果詳見表1。
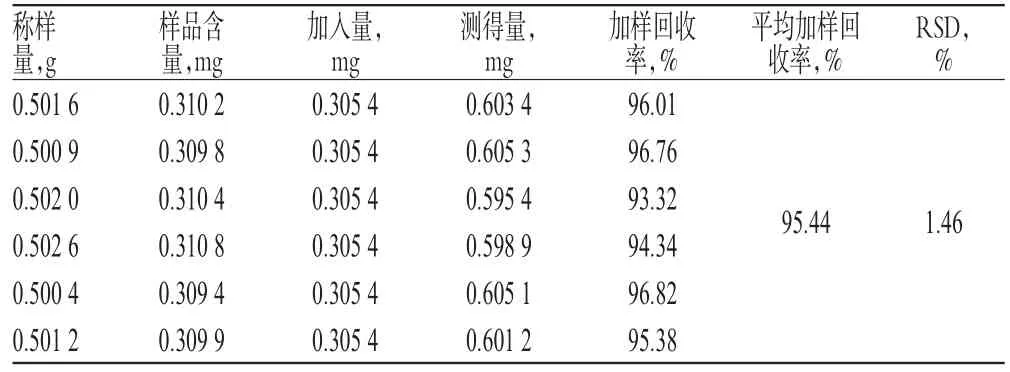
表1 淫羊藿苷加樣回收率試驗結果(n=6)Tab 1 Results of recovery test of icariin(n=6)
2.2.11 淫羊藿苷的含量測定 精密量取黃麥顆粒(批號:20131218)7.5 g,按“2.2.3”項下方法制備供試品溶液,按“2.2.1”項下色譜條件連續進樣3次。結果,樣品中淫羊藿苷的含量分別為 600.0、624.3、630.4μg/g(n=3),平均(618.4±15.9)μg/g。
2.3 金絲桃苷及二苯乙烯苷的含量測定
2.3.1 色譜條件色譜柱:Waters XBridgeTMC18(250mm×4.6mm,5 μm);流動相:0.1%醋酸溶液-乙腈(85∶15,V/V);流速:1.0ml/min;檢測波長:360nm(金絲桃苷)、320nm(二苯乙烯苷);柱溫:室溫;進樣量:5μl。
2.3.2 對照品溶液的制備 精密稱取金絲桃苷對照品適量,用稀乙醇逐級稀釋成12.3、24.6、49.2、98.4、196.8μg/ml的對照品溶液,避光保存;精密稱取二苯乙烯苷對照品適量,用稀乙醇逐級稀釋成12.64、25.28、50.56、101.2、202.4μg/ml的對照品溶液,避光保存。
2.3.3 供試品溶液的制備 取黃麥顆粒(批號:20131218)13.5 g,加入蒸餾水150ml攪拌均勻,于70℃水浴30min,放冷,用濾紙濾過。取濾液20ml,用乙酸乙酯60、40、40ml萃取3次,靜置過夜;合并乙酸乙酯層,于80℃水浴中蒸干,殘渣用稀乙醇定容至10ml,經0.22 μm微孔濾膜濾過,取續濾液,即得,避光保存。
2.3.4 陰性對照溶液的制備 按照黃麥顆粒處方比例,分別制備不含菟絲子及何首烏的陰性樣品,分別精密稱取7.5 g,按“2.3.3”項下方法操作,即得。
2.3.5 專屬性試驗 在“2.3.1”項色譜條件下,金絲桃苷、二苯乙烯苷和其他組分色譜峰基本分離;同時取“2.3.4”項下陰性對照溶液進樣。結果,陰性對照溶液在金絲桃苷及二苯乙烯苷色譜峰位置處無相應峰出現。色譜見圖3。
2.3.6 線性關系考察 精密量取不同質量濃度的金絲桃苷對照品溶液12.3、24.6、49.2、98.4、196.8μg/ml各5μl,注入HPLC儀,按“2.3.1”項下色譜條件進樣測定,記錄金絲桃苷峰面積。以金絲桃苷質量濃度(x,μg/ml)為橫坐標、峰面積(y)為縱坐標進行線性回歸,得回歸方程為y=4.171×10-5x+5.6882(r=0.9999)。結果表明,金絲桃苷質量濃度在12.3~196.8μg/ml范圍內與其峰面積積分值呈良好的線性關系;精密量取不同質量濃度的二苯乙烯苷對照品溶液12.64、25.28、50.55、101.1、202.2μg/ml各5μl,注入HPLC儀,按“2.3.1”項下色譜條件進樣測定,記錄二苯乙烯苷峰面積。以二苯乙烯苷質量濃度(x,μg/ml)為橫坐標、峰面積(y)為縱坐標進行線性回歸,得回歸方程為y=3.900×10-5x+6.9674(r=0.9998)。結果表明,二苯乙烯苷質量濃度在12.64~202.2μg/ml范圍內與其峰面積積分值呈良好的線性關系。

圖3 金絲桃苷及二苯乙烯苷的高效液相色譜圖A.金絲桃苷對照品(49.2μg/ml);B.供試品(波長=360nm);C.缺菟絲子的陰性對照;D.二苯乙烯苷對照品(50.55μg/ml);E.供試品(波長=320nm);F.缺何首烏的陰性對照;1.金絲桃苷;2.二苯乙烯苷Fig 3 HPLC Chromatograms of hyperin and stilbene glucosideA.control substance of hyperin(49.2μg/ml);B.test sample(control substance wavelength=360nm);C.negative solution without China dodder;D.control substance of stilbene glucoside(50.55μg/ml);E.test sample(wavelength=320nm);F.negative solution without Fallopia multiflora;1.hyperin;2.stilbene glucoside
2.3.7 精密度試驗 分別吸取“2.3.2”項下質量濃度為24.6μg/ml的金絲桃苷對照品溶液及25.28μg/ml的二苯乙烯苷對照品溶液各5μl,按“2.3.1”項下色譜條件分別連續進樣6次。結果,金絲桃苷、二苯乙烯苷的RSD分別為1.67%、1.23%(n=6),表明儀器精密度良好。
2.3.8 穩定性試驗 取批號為20131218的黃麥顆粒13.5 g,按“2.3.3”項下方法制備供試品溶液,分別在0、2、4、8、12、24h進樣測定。結果,金絲桃苷、二苯乙烯苷的RSD分別為1.74%、2.44%(n=6),說明供試品溶液在24h內穩定性良好。
2.3.9 重復性試驗 取批號為20131218的黃麥顆粒適量,按“2.3.3”項下方法制備得到6份供試品溶液,吸取5μl,按“2.3.1”項下色譜條件進樣測定。結果,金絲桃苷、二苯乙烯苷的RSD分別為2.15%、2.92%(n=6),表明本方法重復性良好。
2.3.10 加樣回收率試驗 精密稱取黃麥顆粒樣品(批號:20131218)6份,分別精密加入金絲桃苷及二苯乙烯苷對照品各適量,按“2.3.3”項下方法制備供試品溶液,再按“2.3.1”項下色譜條件進樣測定,記錄峰面積,并計算加樣回收率,結果分別見表2、表3。
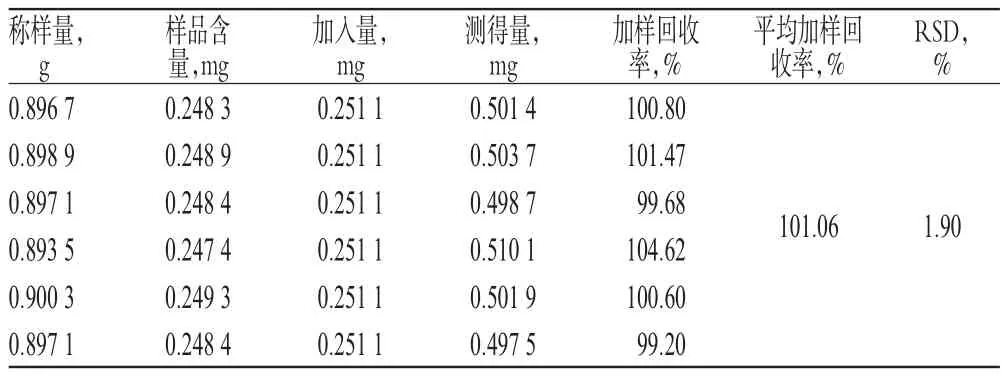
表2 金絲桃苷加樣回收率試驗結果(n=6)Tab 2 Results of recovery test of hyperin(n=6)
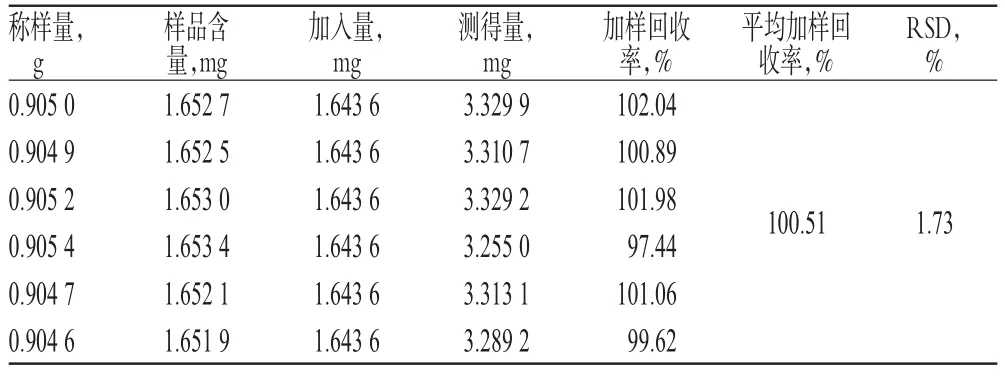
表3 二苯乙烯苷加樣回收率試驗結果(n=6)Tab 3 Results of recovery test of stilbene glucoside(n=6)
2.3.11 金絲桃苷和二苯乙烯苷的含量測定 精密量取黃麥顆粒(批號:20131218)13.5 g,按“2.3.3”項下方法制備供試品溶液,按“2.3.1”項下色譜條件連續進樣3次。結果,樣品中金絲桃苷的含量分別為 276.9、280.2、286.5μg/g(n=3),平均(276.9±4.9)μg/g;二苯乙烯苷的含量分別為1806.3、1843.9、1828.3μg/g(n=3),平均(1826.2±18.9)μg/g。
3 討論
黃麥顆粒是根據我院院內經典制劑黃麥合劑進行劑型改造之后的中成藥,故薄層色譜及高效液相色譜法可借鑒黃麥合劑中對各個成分的定性及定量分析[1],但由于顆粒劑中添加有部分輔料,前處理過程仍有所不同。
3.1 洗滌次數及濃度的選擇
文獻報道,乙酸乙酯萃取淫羊藿苷樣品較為常用[8],而萃取過程中需要使用碳酸鈉溶液進行洗滌[9],以去除雜峰的影響。筆者使用了與黃麥合劑中相同的洗滌方法后發現,峰面積較小,加樣回收率均不足85%,故對洗滌次數(1、2次)及Na2CO3溶液的濃度(5%、2.5%)進行了考察。結果顯示,2.5%Na2CO3溶液洗滌1次后,峰面積明顯大于5%Na2CO3溶液洗滌2次,且雜質峰對淫羊藿苷峰無干擾,完全能夠滿足2010年版《中國藥典》(一部)[10]要求,故最終決定使用2.5%Na2CO3溶液洗滌1次。
3.2 避光操作
在測定二苯乙烯苷過程中需全程避光操作。預試驗由于沒有采取避光措施,二苯乙烯苷濃度偏小,且重復性較差(RSD=5.21%,n=6);采用避光措施之后,峰面積顯著提高,重復性也明顯改善(RSD=2.92%,n=6)。
3.3 雙波長檢測色譜條件的選擇
在考察黃麥顆粒中主要成分的HPLC檢測方法時,利用Waters 2487型紫外檢測器的雙波長功能,將金絲桃苷與二苯乙烯苷同時進行測定(金絲桃苷檢測波長=360nm、二苯乙烯苷檢測波長=320nm)。考慮到金絲桃苷分離度要求較高,選擇了Waters XBridgeTMC18色譜柱(250mm×4.6mm,5 μm)。在金絲桃苷與二苯乙烯苷的溶劑選擇上進行了預試驗,分別用甲醇、無水乙醇、稀乙醇定溶后檢測。結果,稀乙醇中金絲桃苷與二苯乙烯苷的峰面積較大,且峰型好,故最終選擇用稀乙醇溶解。流動相比例的選擇在前期研究的基礎上[11],參照相關文獻[12]也進行了預試驗,分別將流動相的體積比例調整為81∶19、82∶18、85∶15后進樣。結果,在85∶15的情況下,樣品中金絲桃苷與二苯乙烯苷周圍的雜質峰均較少,分離度高,且保留時間接近(金絲桃苷約為22min、二苯乙烯苷約為24min)。故最終選定為0.1%醋酸溶液-乙腈(85∶15,V/V)。選擇0.1%醋酸溶液主要為了保證在改善金絲桃苷的峰型的情況下,不影響二苯乙烯苷的峰型。
綜上所述,本方法操作簡便、重復性好、結果準確可靠,可用于黃麥顆粒的質量控制。
[1]沈金芳,孫黎.黃麥合劑中制何首烏與黃芪的鑒別及二苯乙烯苷的含量測定[J].藥學服務與研究,2009,9(4):289.
[2]曾慶岳,王云山.淫羊藿藥理作用研究進展[J].醫藥導報,2012,31(4):462.
[3]李淑芳.中藥黃芪藥理作用研究進展[J].湖北中醫雜志,2013,35(6):73.
[4]李洪兵.何首烏的現代藥理學研究綜述[J].云南中醫中藥雜志,2012,33(6):72.
[5]張偉,陳素紅,呂圭源.菟絲子功效性味歸經與現代藥理學的相關性研究[J].時珍國醫國藥,2010,21(4):808.
[6]林曉,周強峰,徐德生.麥冬藥理作用研究進展[J].上海中醫藥雜志,2004,38(6):59.
[7]王鴻祥,陳斌,胡凱,等.黃麥合劑治療脾腎陽虛少和弱精癥33例[J].醫藥導報,2011,30(1):24.
[8]葉咸鈺,楊水新,曹恒斌.HPLC測定強筋合劑中淫羊藿苷的含量[J].浙江中醫藥大學學報,2010,34(6):920.
[9]蔡鴻飛,高幼衡,劉軍,等.HPLC法測定前列舒樂顆粒中淫羊藿苷的含量[J].中國新藥與臨床藥理,2008,19(4):307.
[10]國家藥典委員會.中華人民共和國藥典:一部[S].2010年版.北京:中國醫藥科技出版社,2010:附錄130、ⅥB.
[11]楊娟,金冠欽,孫黎,等.HPLC法測定黃麥合劑中金絲桃苷的含量[J].中國藥師,2012,15(12):1739.
[12]鄭清明,秦路平,鄭漢臣,等.國產11種金絲桃屬植物中化學成分的含量分析[J].第二軍醫大學學報,2003,24(4):457.
