復方王不留行片的質量標準研究
2015-05-21 08:55:54趙建穎盧曉梅李亮亮許明哲洛陽市食品藥品檢驗所河南洛陽471023
中國藥房 2015年24期
關鍵詞:黃酮
趙建穎,盧曉梅,李亮亮,許明哲(洛陽市食品藥品檢驗所,河南 洛陽 471023)
復方王不留行片是由王不留行、鄰氨基苯甲酸、干酵母、乳酸鈣組成的復方制劑,具有增強體質、通經活血、散結、下乳、消腫、消癰的功效,臨床上用于產后氣血虧損、乳汁不通不下或乳少及乳痛、乳腫等癥。國家藥品標準化學藥品地方標準上升國家標準第十四冊WS-10001-(HD-1369)-2003未對方中主藥王不留行進行質量控制,文獻報道[1]僅見方中王不留行薄層色譜(TLC)鑒別方法,未見王不留行中王不留行黃酮苷的含量測定方法。為了有效地控制產品質量,筆者建立了TLC法對王不留行進行定性鑒別,采用高效液相色譜(HPLC)法同時測定方中鄰氨基苯甲酸及王不留行黃酮苷的含量。
1 材料
1.1 儀器
U3000型HPLC儀,包括LPG-3400SD型泵、WPS-3000SL型自動進樣器、TCC-3000RS型柱溫箱、VWD-3100型紫外檢測器、Chromeleon色譜工作站(美國Dionex公司);92SM-202-A-DR電子分析天平(瑞士Precisa公司);UV-2401PC型紫外-可見分光光度計(日本島津公司);KQ5200DE型數控超聲波清洗器(昆山市超聲儀器有限公司)。
1.2 藥品與試劑
復方王不留行片(白云山湯陰東泰藥業有限責任公司,批號:130302、120606、130311、131104;焦作市博愛藥業有限公司,批號:110621)。王不留行黃酮苷對照品(批號:111853-201001,純度:91.7%)、王不留行對照藥材(批號:121094-200703)均購自中國食品藥品檢定研究院;鄰氨基苯甲酸對照品(批號:C1305010,純度:99.5%)購自上海晶純生化科技股份有限公司;聚酰胺薄膜(浙江省臺州市路橋四甲生化塑料廠);甲醇為色譜純,磷酸、乙醇、無水乙醇、三氯化鋁均為分析純,水為娃哈哈純凈水。
2 方法與結果
2.1 王不留行的TLC鑒別
取本品,除去糖衣,粉碎,取粉末適量(約相當于王不留行1.0 g),置于錐形瓶中,加70%甲醇40ml,超聲(功率:200 W,頻率:40 kHz)處理30min,放冷,濾過,濾液作為供試品溶液。另取王不留行對照藥材1 g,照上述供試品溶液的制備方法制成對照藥材溶液。再取王不留行黃酮苷對照品適量,加甲醇制成每1ml含0.1mg的溶液,作為對照品溶液。再取“2.2.2”項下缺王不留行的陰性樣品適量,照上述供試品溶液的制備方法制成缺王不留行的陰性樣品溶液。按TLC法[2]試驗,吸取上述4種溶液各2μl,分別點于同一聚酰胺薄膜上,以無水乙醇-水(4∶6,V/V)為展開劑,展開,取出,晾干,采用2%三氯化鋁乙醇溶液浸漬法顯色,熱風吹干,置紫外光燈(365nm)下檢視。結果,供試品溶液中,在與對照藥材色譜和對照品色譜相應的位置上,顯相同顏色的熒光斑點,缺王不留行的陰性樣品無干擾,詳見圖1。

圖1 薄層色譜圖1~3、7~8.5批樣品;4.王不留行黃酮苷對照品;5.王不留行對照藥材;6.缺王不留行的陰性樣品Fig 1 TLC chromatograms1-3,7-8.test sample;4.reference substance of vaccarin;5.reference substance of Vaccaria segetalis;6.negative sample without V.segetalis
2.2 含量測定
2.2.1 色譜條件色譜柱:YMC-Pack Pro C18(250mm×4.6mm,5 μm);流動相:甲醇為流動相A,0.3%磷酸溶液為流動相B,梯度洗脫,程序詳見表1;流速:1.0ml/min;檢測波長:280nm;柱溫:35 ℃;進樣量:10μl。
2.2.2 溶液的制備 (1)對照品溶液。精密稱取王不留行黃酮苷對照品30.38mg,置于50ml量瓶中,用70%甲醇使溶解并稀釋至刻度,搖勻,作為王不留行黃酮苷對照品貯備液。再精密稱取鄰氨基苯甲酸101.08mg,置于50ml量瓶中,再精密量取上述王不留行黃酮苷對照品貯備液5ml,置于同一50ml量瓶中,用70%甲醇稀釋至刻度,搖勻,即得混合對照品溶液。(2)供試品溶液。取樣品40片,除去糖衣,精密稱定,研細,精密稱取適量(約相當于王不留行1.0 g),置于50ml量瓶中,加70%甲醇30ml,超聲(功率:200 W;頻率:40 kHz)處理30min,放置至室溫,用70%甲醇稀釋至刻度,搖勻,即得供試品溶液。(3)陰性對照溶液。按處方比例分別稱取除鄰氨基苯甲酸、王不留行外的其余各成分,按制備工藝分別制成缺鄰氨基苯甲酸、王不留行的陰性樣品,按上述供試品溶液制備方法,制成缺鄰氨基苯甲酸、王不留行的陰性對照溶液。
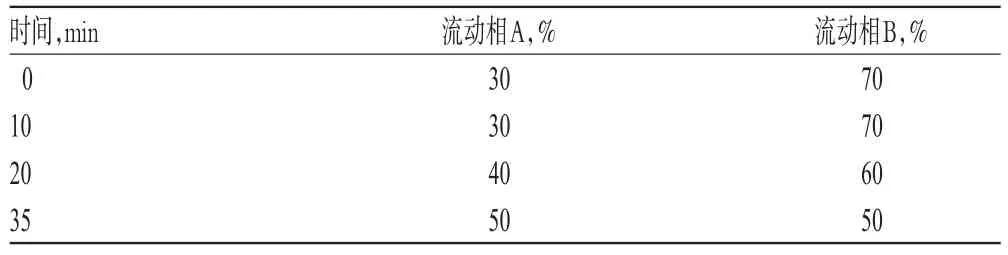
表1 梯度洗脫程序Tab 1 Program of the linear gradient elution
2.2.3 系統適用性試驗 精密吸取“2.2.2”項下的對照品溶液、供試品溶液和陰性對照溶液各10μl,按“2.2.1”項下色譜條件進樣測定,記錄色譜圖。結果,供試品溶液中鄰氨基苯甲酸和王不留行黃酮苷能達到基線分離,理論板數以王不留行黃酮苷計不低于5500,分離度>9.2;陰性對照溶液無干擾。色譜見圖2。

圖2 高效液相色譜圖A.混合對照品;B.供試品;C.缺王不留行的陰性對照;D.缺鄰氨基苯甲酸的陰性對照;1.鄰氨基苯甲酸;2.王不留行黃酮苷Fig 2 HPLC chromatogramsA.mixed reference;B.test sample;C.negative sample without V.segetalis;D.negative sample without anthranilic acid;1.anthranilic acid;2.vaccarin
2.2.4 線性關系考察 分別精密吸取“2.2.2”項下混合對照品溶液1、3、5、7、10、20μl,按“2.2.1”項下色譜條件進樣測定,記錄色譜峰面積。以進樣量(x,μg)為橫坐標、峰面積(y)為縱坐標進行線性回歸,得回歸方程為y王不留行黃酮苷=21.56x-0.100(r=0.9999);y鄰氨基苯甲酸=3.755x+1.296(r=0.9999)。結果表明,王不留行黃酮苷和鄰氨基苯甲酸的進樣量分別在0.0557~2.2287、2.0114~40.2279μg范圍內與各自峰面積呈良好的線性關系。
2.2.5 精密度試驗 精密吸取對照品溶液10μl,按“2.2.1”項下色譜條件重復進樣6次。結果,王不留行黃酮苷和鄰氨基苯甲酸的峰面積的RSD分別為0.97%、0.38%,表明儀器精密度良好。
2.2.6 穩定性試驗 取供試品(批號:130302)溶液適量,分別于配制0、2、6、8、12h時按“2.2.1”項下色譜條件進樣測定,記錄色譜圖。結果,王不留行黃酮苷和鄰氨基苯甲酸峰面積的RSD分別為0.86%、0.79%(n=6),表明供試品溶液12h內穩定性良好。
2.2.7 重復性試驗 取同一批樣品(批號:130302)適量,按“2.2.2”項下方法平行制備6份供試品溶液,按“2.2.1”項下色譜條件進樣測定。結果,樣品中王不留行黃酮苷和鄰氨基苯甲酸的平均含量分別為1.0102、32.016mg/g,RSD分別為1.01%、0.68%,表明該方法重復性良好。
2.2.8 加樣回收率試驗 精密稱取已知含量的樣品(批號:130302)細粉適量(約相當于王不留行0.5 g),共6份,分別置于50ml量瓶中,分別加入鄰氨基苯甲酸對照品約50mg、“2.2.2”項下王不留行黃酮苷對照品貯備液3.0ml,加70%甲醇30ml,按“2.2.2”項下方法制成供試品溶液,按“2.2.1”項下色譜條件測定并計算加樣回收率,結果詳見表2。

表2 加樣回收率試驗結果(n=6)Tab 2 Results of recovery tests(n=6)
2.2.9 樣品含量測定 取5批樣品各適量,按“2.2.2”項下方法制備供試品溶液,再按“2.2.1”項下色譜條進行測定,以外標法計算,結果詳見表3。

表3 樣品含量測定結果(n=3)Tab 3 Results of content determination of samples(n=3)
3 討論
3.1 檢測波長的選擇
精密稱取王不留行黃酮苷和鄰氨基苯甲酸對照品各適量,用70%甲醇配制成適宜濃度的溶液,照紫外-可見分光光度法在200~400nm范圍內掃描吸收光譜。結果表明,王不留行黃酮苷在280nm波長處有最大吸收,鄰氨基苯甲酸在217、243nm波長處均有最大吸收。由于處方中王不留行黃酮苷的含量相對較低,為使兩種成分都有較好的響應,故選擇280nm作為檢測波長。
3.2 流動相的篩選
參考相關文獻[1-8],本試驗比較了多種流動相系統,分離效果均不理想。最終選用甲醇-0.3%磷酸溶液作為流動相,通過優化,使樣品中鄰氨基苯甲酸與王不留行黃酮苷色兩組分達到基線分離,陰性對照樣品無干擾,且各組分分離度良好,峰形佳。
3.3 耐用性考察
本文考察了YMC-Pack Pro C18(250mm×4.6mm,5 μm)、ZORBAX Eclipse XDB-C18(250mm×4.6mm,5μm)、Thermo Syncronis C18(150mm×4.6mm,5 μm)等色譜柱對目標化合物分離度和含量測定結果的影響。結果表明,3個不同廠家的同類型色譜柱對樣品含量測定結果沒有顯著影響,含量測定RSD<2.8%,分離度均>3.4。
3.4 TLC鑒別中展開系統和溫度的考察
筆者曾采用甲醇-水(4∶6,V/V)、甲醇-水(1∶1,V/V)等展開系統對方中王不留行進行TLC鑒別,但陰性對照樣品均有干擾,效果不佳。最終采用無水乙醇-水(4∶6,V/V)的展開系統,該方法陰性對照樣品無干擾,斑點清晰,分離度好。試驗中筆者還發現,溫度對樣品的分離度有較大影響,溫度高時陰性樣品干擾大,斑點分離度差,因此TLC鑒別時應控制溫度在20℃以下。
3.5 含量測定結果分析
由本研究結果可知,不同廠家樣品中王不留行黃酮苷的含量相差極大,且低于2010年版《中國藥典》(一部)[1]中對王不留行藥材最低限量(0.40%)的規定。分析原因,主要是由于原質量標準中無王不留行質量控制,廠家投料用飲片含量差異所致。為了更好地控制復方王不留行片的質量,保證用藥安全、有效,有必要建立王不留行黃酮苷的含量控制標準。
綜上所述,該方法操作簡便、快捷,結果準確、重復性好,可作為復方王不留行片的質量控制方法。
[1]國家藥典委員會.中華人民共和國藥典:一部[S].2010年版.北京:中國醫藥科技出版社,2010:49、附錄ⅥB.
[2]王玉霞,殷永偉,耿琴.HPLC測定腎石通顆粒中王不留行黃酮苷的含量[J].數理醫藥學雜志,2012,25(5):557.
[3]韓晉,李娜,劉冬,等.復方靈芝乳膏中王不留行黃酮苷含量測定方法研究[J].解放軍藥學學報,2012,28(5):415.
[4]孟賀,陳玉平,秦文杰,等.HPLC測定王不留行中王不留行黃酮苷的含量[J].中國中藥雜志,2010,35(16):2072.
[5]張輝,陳乃江.HPLC法測定復方王不留行片中鄰氨基苯甲酸的含量[J].現代中藥研究與實踐,2009,23(4):63.
[6]王小雪,鄭文捷,謝國祥,等.高效毛細管電泳同時測定板藍根中水楊酸、丁香酸、苯甲酸和鄰氨基苯甲酸[J].中國中藥雜志,2009,34(2):189.
[7]方建國,萬進,湯杰,等.大孔吸附樹脂分離純化大青葉有機酸部位的實驗研究[J].中國藥房,2007,18(16):1214.
[8]王文清,張飛,方建國,等.反相高效液相色譜法測定大青葉中鄰氨基苯甲酸與丁香酸的含量[J].醫藥導報,2005,25(5):456.
猜你喜歡
四川蠶業(2021年2期)2021-03-09 03:15:32
四川蠶業(2021年3期)2021-02-12 02:38:46
中成藥(2018年11期)2018-11-24 02:57:00
中成藥(2017年8期)2017-11-22 03:19:40
中成藥(2017年10期)2017-11-16 00:50:13
中成藥(2017年4期)2017-05-17 06:09:50
哈爾濱醫藥(2016年1期)2017-01-15 13:43:16
天然產物研究與開發(2016年11期)2016-06-15 20:29:17
湖南師范大學自然科學學報(2015年1期)2015-02-27 14:50:04
安徽醫藥(2014年12期)2014-03-20 13:15:15
