HPLC法測定藏藥仁青芒覺膠囊中西紅花苷-I和西紅花苷-Ⅱ的含量
2015-06-07 05:55:36楊博涵朱旭江
中國民族民間醫藥 2015年20期
關鍵詞:方法
楊博涵朱旭江
1.天津市醫藥科學研究所,天津 300020;2.甘肅省藥品檢驗研究院,甘肅 蘭州 730070
HPLC法測定藏藥仁青芒覺膠囊中西紅花苷-I和西紅花苷-Ⅱ的含量
楊博涵1朱旭江2*
1.天津市醫藥科學研究所,天津 300020;2.甘肅省藥品檢驗研究院,甘肅 蘭州 730070
目的:建立仁青芒覺膠囊中西紅花的含量測定方法。方法:采用HPLC法,用光電二極管陣列檢測器 (DAD),流動相為甲醇-水(50∶50),流速1.0 ml/min,檢測波長440nm,柱溫25℃,對西紅花苷-Ⅰ、西紅花苷-Ⅱ進行定量測定。結果:西紅花苷-Ⅰ在0.0336~0.4987μg范圍內呈良好的線性關系,r=0.9999,加樣回收率為97.43%,RSD=2.18%;西紅花苷-Ⅱ在0.0129~0.1818μg范圍內呈良好的線性關系,r=0.9998,加樣回收率為99.75%,RSD=1.53%結論:該方法操作簡便、專屬性、重現性好,可有效地控制仁青芒覺膠囊的質量。
仁青芒覺膠囊;HPLC;西紅花苷-Ⅰ;西紅花苷-Ⅱ
仁青芒覺始載于 《盤德瓊乃》,是藏族驗方。仁青芒覺膠囊由毛訶子、蒲桃、西紅花、木香、丁香、朱砂、馬錢子等藥材組成的復方制劑,具有清熱解毒,益肝養胃,明目醒神,愈瘡,滋補強身的功效。其現行標準為國家食品藥品監督管理局標準(YBZ07062005-2010Z),含量測定項為檢查沒食子酸的含量,但處方中多味藥材含有沒食子酸,故該法的專屬性不強。仁青芒覺膠囊處方組成比較復雜,含量測定存在一定難度,導致該藥含量測定研究報道較少[1-3]。處方中的西紅花為鳶尾科植物番紅花 (Crocus sativus L.)的干燥柱頭,系貴重藥材,具有活血化瘀,清熱解毒,解郁安神的作用,且仁青芒覺膠囊為保密處方,處方量不明確。為了保證用藥的有效性,試驗采用高效液相色譜法建立了仁青芒覺膠囊中西紅花苷-I和西紅花苷-Ⅱ的含量測定方法,該法操作簡便、結果準確,可用于產品內在質量的控制。
1 儀器與試藥
1.1 儀器 Waters e2695高效液相色譜儀(Waters 2998 DAD檢測器,Empower色譜工作站,美國Waters公司);Mettler AE 240電子天平(瑞士梅特勒);Sartorius R200D電子天平(德國賽多利斯公司);KH-500DE超聲波清洗器(昆山禾創超聲儀器有限公司);HGC-24A氮吹儀(天津市恒奧科技發展有限公司)。
1.2 試藥 西紅花苷-I(批號:111588-200501)和西紅花苷-Ⅱ(批號:110589-201304,含量92.4%)均購自中國食品藥品檢定研究院;仁青芒覺膠囊 (批號:100501、120001、130101)由甘肅省某藏藥企業提供。甲醇 (色譜純,德國默克Merck公司);水為超純水,其余試劑均為分析純。
2 方法
2.1 色譜條件 色譜柱:資生堂CAPCELL PAK C18(4.6 mm×250mm,5μm),Waters Symmetry C18(4.6mm× 250mm,5μm),Thermo scientific BDS HYPERSIL C18(4.6mm×250mm,5μm);流動相:甲醇-水(50:50);流速:1ml/min;柱溫:25℃;檢測波長:440nm。
2.2 溶液的制備
2.2.1 對照品溶液的制備 精密稱取西紅花苷-Ⅰ對照品8.28mg,至50ml棕色量瓶中,加稀乙醇稀釋至刻度,作為西紅花苷-Ⅰ對照品儲備液。精密稱取西紅花苷-Ⅱ對照品8.20mg,至50ml棕色量瓶中,加稀乙醇稀釋至刻度,作為西紅花苷-Ⅱ對照品儲備液。分別精密吸取西紅花苷-Ⅰ對照品儲備液5ml和西紅花苷-Ⅱ對照品儲備液2ml至50ml容量瓶中,加稀乙醇稀釋至刻度,制成混合對照品溶液。
2.2.2 供試品溶液的制備 取裝量差異項下的本品內容物,研細,取約0.5g,精密稱定,置50ml棕色量瓶中,加稀乙醇適量,置冰浴中超聲處理 (功率200 W,頻率60KHz)30min,放至室溫,加稀乙醇稀釋至刻度,搖勻,濾過,取續濾液,即得。
2.2.3 陰性供試品的制備 按照供試品溶液的制備方法,分別制備缺西紅花的陰性供試品溶液。
2.2.4 樣品的測定 按“2.1”項下色譜條件,分別精密吸取對照品溶液、供試品溶液及陰性供試品溶液各5~10μl,注入液相色譜儀,測得液相色譜圖,結果見圖1。結果表明,西紅花苷-Ⅰ、西紅花苷-Ⅱ色譜分離良好,且陰性對測定結果無干擾。
2.3 方法學考察
2.3.1 線性關系的考察 取“2.2.1”項下混合對照品溶液,按“2.1”項的色譜條件,分別進樣2、4、8、10、14、18、22、26、30μl,測定西紅花苷-Ⅰ、西紅花苷-Ⅱ的峰面積,以進樣對照品質量 (μg)為橫坐標 (X),峰面積為縱坐標 (Y),繪制標準曲線,計算回歸方程,結果見表1。結果表明,西紅花苷-Ⅰ和西紅花苷-Ⅱ在線性范圍內呈良好的線性關系。
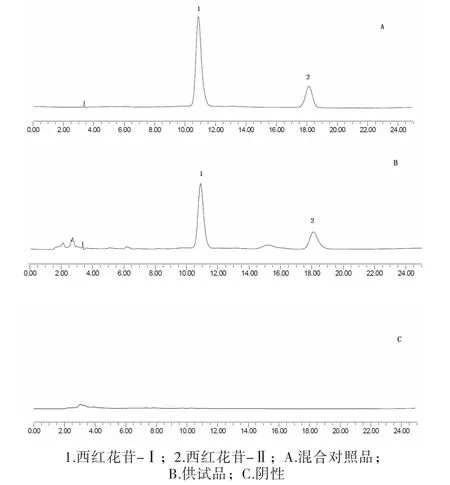
圖1 高效液相色譜圖

表1 西紅花苷Ⅰ和西紅花苷Ⅱ的線性回歸方程
2.3.2 精密度實驗 精密吸取同一份供試品溶液10μl,注入液相色譜儀,連續進樣6次,測定峰面積積分值,西紅花苷-Ⅰ和西紅花苷-Ⅱ的RSD分別為0.5%和0.4%,結果表明進樣精密度良好。
2.3.3 穩定性試驗 取同一供試品溶液(批號:130001)分別在0、2、4、6、8、24 h,按“2.1”項的色譜條件各進樣1次,記錄峰面積積分值,西紅花苷-Ⅰ和西紅花苷-Ⅱ的RSD分別為1.4和1.3%,表明供試品溶液在24h內基本穩定。
2.3.4 重復性試驗 取同一批供試品(批號:130001)6份,按“2.2.2”項下方法處理,“2.1”項的色譜條件測定含量。結果西紅花苷-Ⅰ和西紅花苷-Ⅱ的RSD分別為0.98%和1.02%。
2.3.5 加樣回收率 精密量取西紅花苷-Ⅰ對照品儲備液(0.1656mg/ml)1.5、2.0、2.5ml各3份和西紅花苷-Ⅱ對照品儲備液(0.06061mg/ml)0.8、1.0、1.2ml各3份,置50ml棕色量瓶中,溶劑采用氮吹儀吹干,再分別精密稱取已知含量同一批號(批號:130001,西紅花苷-Ⅰ含量1.2605 mg/g;西紅花苷-Ⅱ含量0.4376 mg/g)的供試品9份,按“2.2.2”項下方法制備供試品溶液,依法測定,計算西紅花苷-Ⅰ和西紅花苷-Ⅱ的回收率。結果見表2、表3。
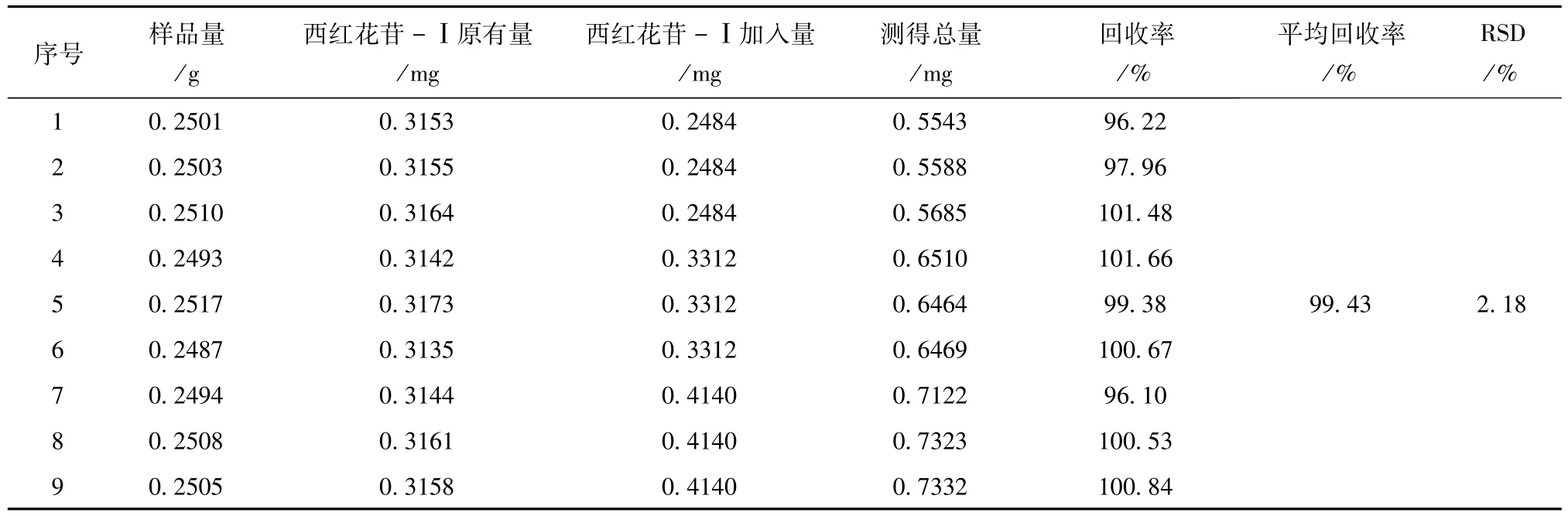
表2 西紅花苷-Ⅰ加樣回收率
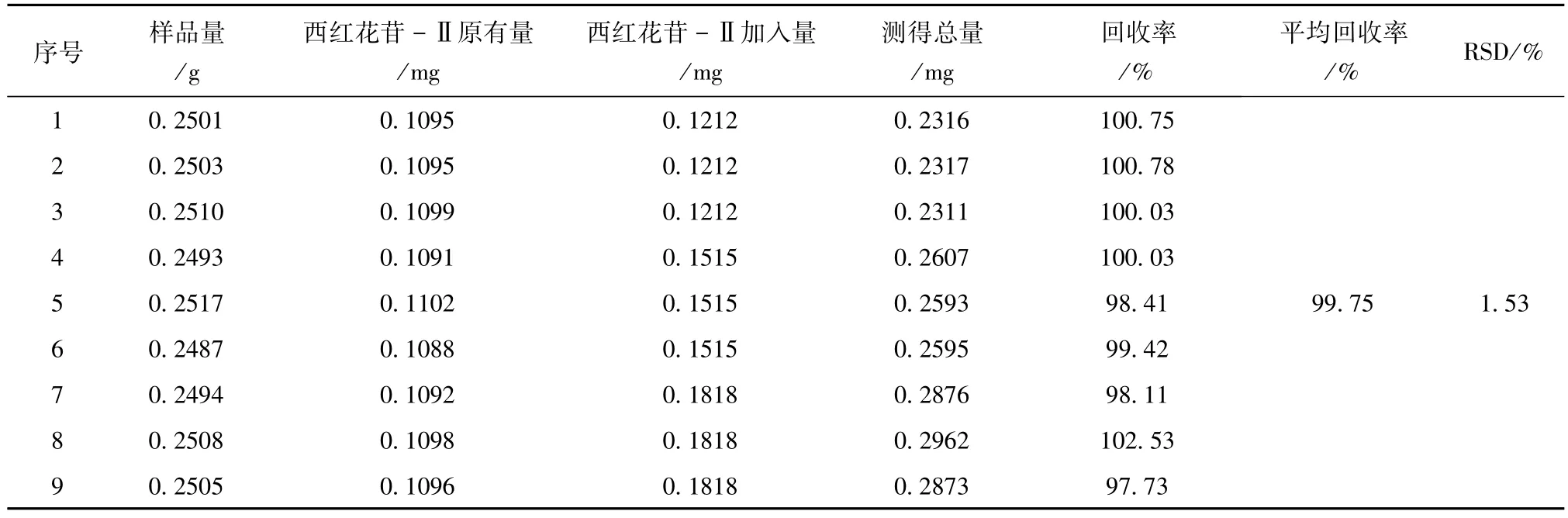
表3 西紅花苷-Ⅱ加樣回收率
2.4 樣品測定結果 按供試品溶液的制備方法和測定方法,對樣品進行制備和測定,分別精密吸取對照品溶液與供試品溶液各10μl,按“2.1”項下色譜條件進樣分析,按外標法以峰面積計算樣品中西紅花苷-Ⅰ和西紅花苷-Ⅱ含量,測定3批供試品的含量,結果見表4。

表4 仁青芒覺膠囊含量測定結果
3 討論
3.1 提取方法的選擇 參考中國藥典[4]及有關文獻[5-6],提取溶劑選定為稀乙醇。西紅花苷文獻報道較多[7-8],大多采用超聲提取方法。西紅花苷為水溶性類胡蘿卜素類成分,分子中具有多個共軛雙鍵,導致其穩定性差,且對溫度非常敏感[9]。研究對供試品在冰浴中和常溫下超聲處理方法進行了對比研究。結果表明,冰浴比常溫提取所測得的西紅花苷-Ⅰ和西紅花苷-Ⅱ峰面積略高,故采用冰浴超聲提取作為仁青芒覺膠囊中西紅花苷-Ⅰ和西紅花苷-Ⅱ的提取方法。
3.2 超聲時間的選擇 根據預實驗的結果,供試品制備采用冰浴中分別超聲(功率200 W,頻率60 KHz)20、25、30、40、50 min,依法測定峰面積,結果表明,超聲提取30 min時的測定值略高,故選擇提取時間為30 min。
3.3 測定波長的選擇 對西紅花苷-Ⅰ和西紅花苷-Ⅱ進行全波長掃描,結果在440 nm波長處有最大吸收,故選擇440nm作為測定波長。
3.4 方法適用性試驗 取同一批樣品按供試品溶液的制備方法進行制備,分別采用三種品牌的色譜柱測定,西紅花苷-Ⅰ和西紅花苷-Ⅱ測得含量的RSD分別為1.92%和1.56%,結果無明顯差別,說明該方法適用性較好。
3.5 小結 樣品各批次間西紅花苷-Ⅰ和西紅花苷-Ⅱ的含量差異較大,說明現行的質量標準不能控制產品質量,使各批樣品的投料量存在差異。實驗采用HPLC法同時測定仁青芒覺膠囊中西紅花苷-Ⅰ和西紅花苷-Ⅱ的含量,此法方便可靠、重現性好,為仁青芒覺膠囊的質量控制與評價提供了參考資料。
[1]田薇,陳朝暉.薄層色譜法檢查仁青芒覺膠囊中烏頭堿、士的寧的限量[C].甘肅省化學會成立六十周年學術報告會暨二十三屆年會論文集,2003,419.
[2]馬寧,朱旭江,楊錫,等.仁青芒覺膠囊質量標準研究 [J].中國中醫藥信息雜志,2014,21(6):72-75.
[3]房少新,趙利平.藏藥仁青芒覺中微量元素的測定 [J].光譜實驗室,2013,30(2):916-919.
[4]國家藥典委員會.中華人民共和國藥典 (一部)[M].北京:中國醫藥科技出版社,2010.
[5]周素娣,仲惠娟.HPLC測定西紅花提取物及其片劑中各西紅花苷含量[J].藥物分析雜志,1998,18(3):159-162.
[6]李彬,周素娣,周錦祥.中藥西紅花指紋圖譜研究 [J].海峽藥學,2005,17(3):83-85.
[7]尼珍,阿萍,格桑索朗.HPLC法測定藏藥二十五味珍珠丸中西紅花苷Ⅰ的含量[J].藥物分析雜志,2011,31(1):151-153.
[8]繆玉山,黃根林,倪沖,等.反相高效液相色譜法對不同來源西紅花藥材中西紅花苷-1、苷-2的定量分析[J].中國藥學雜志,2002,22(11):654-656.
[9]付小梅,王崢濤.西紅花苷-Ⅰ的穩定性研究[J].食品科學,2012,33(5):71-73.:
HPLC determ ination of crocin-Ⅰand crocin-Ⅱin Renqing M angjue capsules
YANG Bohan1ZHU Xujiang*
1.Tianjin Institute of Medical and Pharmaceutical Sciences,Tianjin 300020,China;2.Gansu Institute for Drug Control,Lanzhou 730070,China
Objective To set up the quality standard of Renqing Mangjue capsules.M ethods The content of crocin-Ⅰand crocin-Ⅱwere determined by HPLC,which conducted with a DAD detector and the detection wavelength set at440nm.The column was C18(4.6mm×250mm,5μm)withmobile phase consisted ofmethanol-water(50:55)and the flow rate was 1.0 ml·min-1. The column temperaturemaintained at 25℃.Results HPLC determined that crocin-Ⅰwas in range from 0.0336 to 0.4987μg,which presented a good linear relationship(r=0.9999).The average recovery was 97.43%,RSD was 2.18%.HPLC determined that crocin-Ⅱwas in range from 0.0129 to 0.1818μg,which presented a good linear relationship(r=0.9998).The average recovery was 99.75%,RSD was1.53%.Conclusion Themethod is simple in operation.The results are accurate,reliable and good in reproducibility.Themethod can effectively control the quality of Renqing Mangjue capsules.
Renqing Mangjue capsules;HPLC;crocin-Ⅰ;crocin-Ⅱ
R284.1
A
1007-8517(2015)20-0016-03
2015.07.10)
楊博涵,女,學士。E-mail:1132563453@qq.com
朱旭江,博士,主任藥師。主要從事藥品質量標準研究、色譜學及相關技術。E-mail:gszhuxujiang@163.com
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
