甲醇或乙醇水蒸氣重整制氫高效新型催化劑的研發
2015-06-24 14:25:33梁雪蓮劉志銘謝建榕陳秉輝林國棟張鴻斌
廈門大學學報(自然科學版) 2015年5期
關鍵詞:催化劑
梁雪蓮,劉志銘,謝建榕,陳秉輝,林國棟,張鴻斌
(廈門大學 化學化工學院,固體表面物理化學國家重點實驗室,醇醚酯化工清潔生產國家工程實驗室,福建廈門361005)

·綜 述·
甲醇或乙醇水蒸氣重整制氫高效新型催化劑的研發
梁雪蓮,劉志銘,謝建榕,陳秉輝,林國棟,張鴻斌*
(廈門大學 化學化工學院,固體表面物理化學國家重點實驗室,醇醚酯化工清潔生產國家工程實驗室,福建廈門361005)
新型替代清潔能源的開發是大勢所趨.氫能作為理想的清潔能源之一,已引起人們廣泛重視.但氫能廣泛地應用,尤其是用作電動車燃料電池的燃料,必須解決其儲存和輸運的技術問題.采用液體作為氫的載體,通過其重整即時產生燃料H2在商業上具有重要實用價值.在諸多可重整制H2的液體燃料中,甲醇、乙醇以其反應溫度和壓力低,H/C 比高,無NOx、SOx排放,并可利用現行動力燃料輸配系統等優點而占據優勢.本文介紹甲醇、乙醇制氫技術的研發動態,重點報道本實驗室在甲醇、乙醇經水蒸氣重整制氫用高效高穩定性催化劑的研發進展.
氫能;甲醇水蒸氣;重整制氫;乙醇水蒸氣
氫是一種理想的清潔燃料,有著化石燃料無法比擬的一些優點,諸如:放熱效率高;在燃燒過程中產生的廢物只有水,用作燃料電池(fuel cell)的燃料時基本上可實現零排放;通用性強,可用于大多數終端燃燒設備;也是一種可再生的循環燃料,能輸運、儲存.長期以來化石燃料的廣泛使用,既對全球環境造成嚴重污染,也難免因過度開采而日漸枯竭.新型替代清潔能源的開發是大勢所趨.氫能作為理想的新型替代能源之一,已引起人們廣泛重視[1-2].
現有制氫方法有多種,諸如:電解水制氫、太陽能制氫、化石燃料制氫、甲醇制氫、生物質制氫、熱化學方法制氫、利用核能制氫等[3-4],但作為大規模廉價制氫的方法仍在研發之中.從全球制氫的發展趨勢看,氫的生產呈現多種方法/途徑并存的格局,其中天然氣水蒸氣轉化和生物質氣化占主導地位.預計到21世紀后期,生物質制氫可期大幅增長、并成為最主要的制氫途徑.利用可再生資源(尤其是生物質和太陽能)制氫是H2生產的發展方向.
由于H2的密度小、易燃,導致氫能的存儲輸送比化石燃料困難得多,氫能的應用須建立在儲氫技術之上.世界各國對氫儲運技術的研究十分重視,并已取得一定的進展[5-6].儲氫方法可分為物理方法和化學方法.物理方法儲氫指的是通過改變溫度和壓力使氫液化、氣化或吸附、脫附,從而達到儲存氫的目的.化學方法儲氫指的是利用氫和儲氫物質在一定條件下反應,將氫儲存起來,改變條件后,又可以釋放出氫,從而達到儲氫的目的,諸如:金屬氫化物儲氫,有機液態氫化物儲氫和無機物儲氫.
多年來人們一直在努力尋找既有較高的能源利用效率又不污染環境的能源利用方式,而燃料電池就是比較理想的發電技術.燃料電池是一種將存在于燃料與氧化劑中的化學能直接轉化為電能的發電裝置,其發電過程不通過熱機轉換,轉換效率高,沒有污染物排放,幾乎沒有噪聲;而以氫為燃料的燃料電池是最有發展前途的發電裝置之一.近年來,電動車領域成為燃料電池應用的主要方向,并取得許多可喜的進展[7-8].
氫作為一種燃料,欲期廣泛地應用,尤其是作為電動車燃料電池的燃料,尚存在一些亟待解決的問題[9-10],包括:1) 現有的制氫工藝能量消耗大,效率低,其技術有待提高;2) 氫密度小,易氣化、著火、爆炸,這給氫的儲存、運輸及使用帶來一些不便,儲氫技術亟待改進;3) 氫燃料“逃逸率”高,即使用真空密封燃料箱,也以每24小時2%的速率“逃逸”,而汽油每月“逃逸率”一般只1%;4) 添加氫燃料既費時又有危險.鑒于氫燃料電池電動車在運行中存在上述若干不足,采用液體作為氫的載體,通過其重整即時產生氫燃料電池所需要的燃料H2在商業上具有重要實用價值.在諸多可重整制氫的液體燃料中,甲醇、乙醇以其反應溫度和壓力低、H/C 比高、無NOx、SOx等排放物,并可利用現行動力燃料輸配系統等優點而占據優勢[10-12].
本文介紹甲醇和乙醇制氫技術的研發動態,重點報道本實驗室在甲醇、乙醇經水蒸氣重整制氫用作高效新型催化劑的研發進展.
1 甲醇制氫研究概況
現有甲醇制氫主要有如下3條途徑:1) 甲醇分解制氫,2) 甲醇部分氧化制氫,3) 甲醇水蒸氣重整(MSR)制氫.
甲醇分解反應是利用合成氣制甲醇的逆反應,反應式為:CH3OH → CO + 2H2,適合于合成甲醇用的催化劑均可用于其分解.在燃料電池電動車上可以利用燃料電池未反應完的廢氣燃燒提供熱量進行甲醇的分解.該法的不足之處是分解氣中含有大量的CO,而CO使燃料電池的Pt電極嚴重中毒,需將CO轉化,且其體積分數須控制在0.01%以下[13],所以需要較大的轉化器.此方法制氫不宜于直接用于燃料電池電動車上.
用氧氣或空氣部分氧化甲醇,在特定條件下可以得到CO2和H2,反應式為:CH3OH + 0.5O2→ CO2+ 2H2.甲醇部分氧化法制氫的優點是反應屬放熱反應,反應速度快,但反應氣中氫的體積分數不高;由于通入空氣氧化時,其中含N2降低了混合氣中H2的體積分數(使其低于50%),不利于燃料電池的正常工作(因為燃料電池要求氫體積分數為50%~100% )[14].
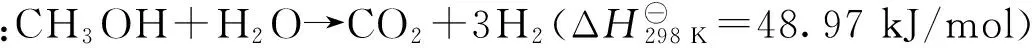
2 MSR制氫催化劑的研發
2.1 研發概況
近20年來,MSR制氫用催化劑的研發一直受到氫能學界的重視[15].Tsai等[16]最早報道用一種準晶態AlCuFe合金微粒作為MSR催化劑,在573 K的反應條件下,H2的產率達到 235 mL/(g·min);高的活性可歸因于該合金催化劑的脆性便于粉碎以獲得較大的表面積;加入Fe的目的在于抑制Cu顆粒的燒結.Liu等[17]報道一種由共沉淀法制備的Ce1-xCuxO2-x復氧化物經還原制得Cu/CeO2作為MSR催化劑,在一種3.9%Cu/CeO2(3.9%為Cu為負載量,即質量分數,下同)上513 K的反應條件下,MSR的轉化率(X)可達53.9%,明顯高于相同Cu載量的Cu/ZnO,Cu/Zn(Al)O 和Cu/Al2O33種催化劑在相同反應條件下的轉化率(分別為37.9%,32.3% 和11.2%).該Cu/CeO2催化劑的高活性可歸因于Cu金屬顆粒的高分散度以及Cu+物種被CeO2載體穩定化;但在493和513 K的反應條件下也觀測到該催化劑緩慢失活.Zhang等[18]報道共沉淀型Cu/ZrO2/Al2O3催化劑,其對MSR顯示出比Cu/Al2O3較高的活性和穩定性.在一種ZrO2質量分數為15%的Cu/ZrO2/Al2O3催化劑上,在523 K的反應條件下,甲醇轉化率可達95.0%(摩爾分數),產物H2的選擇性達99.9%(摩爾分數);反應200 h后甲醇轉化率尚維持在90.0%(摩爾分數).X射線衍射(XRD)檢測結果顯示,在Cu/Al2O3中摻入ZrO2不僅顯著地提高Cu組分的分散度,還有助于防止Cu微晶的聚集燒結.從總體上看,Cu基催化劑的MSR活性和選擇性甚佳,但其耐熱性能差,當反應溫度高于573 K時就容易燒結失活[15],實用前景似乎不被看好.
貴金屬基催化劑用于MSR是另一研發領域[19-26].Iwasa等[19]最先報道一系列Pd基催化劑(包括:Pd/SiO2,Pd/A12O3,Pd/La2O3,Pd/Nb2O5,Pd/Nd2O3,Pd/ZrO2,Pd/ZnO和非負載的Pd)用于MSR反應,結果顯示:載體對MSR的反應活性有強烈影響,以ZnO負載的Pd基催化劑(Pd/ZnO)的產物選擇性最佳.Iwasa等[20-21]隨后陸續報道,在較高溫度下進行Pd/ZnO的還原活化能顯著提高其對MSR反應的催化性能,原先金屬Pd的催化功能因高溫還原生成PdZn合金而大為改進,認為在含有PdZn合金的催化劑上,反應中間物甲醛同水有效地起作用,轉化為CO2和H2,而含有金屬Pd的催化劑與此不同,其反應中間物甲醛卻選擇性地分解生成CO和H2;負載在Ga2O3、In2O3上的Pd,以及負載在ZnO、Ga2O3或In2O3上的Pt,對MSR也都顯示出與Pd/ZnO體系相似的催化活性和選擇性;程序升溫還原(TPR)和XRD測量的結果證實,當用H2還原時上述體系都分別生成PdZn,PdGa,PdIn,PtZn,PtGa和PtIn等合金,進一步證實合金化修改了上述體系的催化功能.
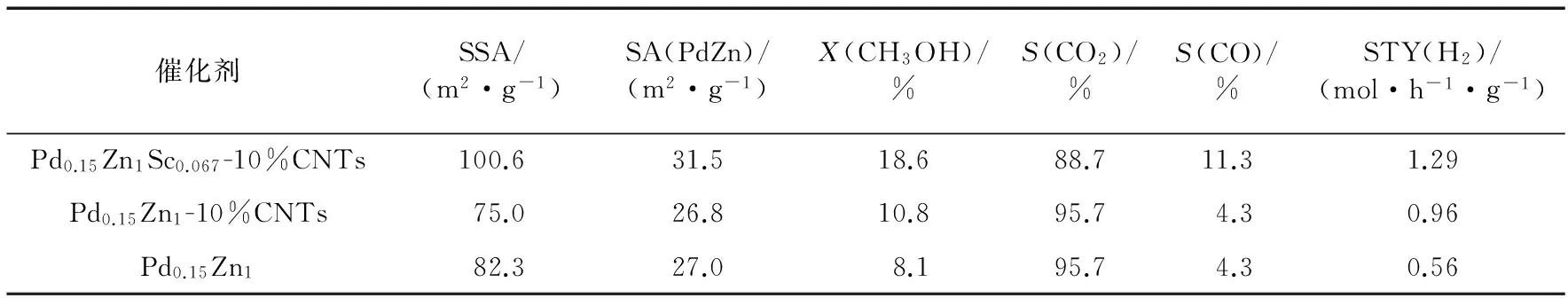
表1 在CNTs和Sc2O3雙促進的Pd-ZnO催化劑及其參比體系上MSR制氫的反應活性[27]
Karim等[23]的對比研究發現,妨礙CO2選擇性提高的,主要是粒徑低于2 nm的金屬Pd顆粒,因而認為高溫H2-還原處理不僅轉化單金屬Pd為PdZn合金,同時還消去粒徑低于2 nm的那些金屬(可能包括合金) 顆粒,這兩個因素都貢獻于CO2選擇性的提高.Chin等[24]也報道過Pd/ZnO對MSR制氫顯示出優良的催化性能,其催化劑制備和預處理研究顯示,Pd/ZnO催化劑對MSR特殊的催化性能可歸因于催化劑的制備和預處理效應.
盡管十多年來在MSR用貴金屬基催化劑的研究開發方面取得了一系列進展,但從工業實用角度考慮,現有一些催化劑的活性,尤其是操作穩定性,仍待改進.
2.2 高效高穩定性催化劑研發
新近本實驗室Yang等[27]研發出一種多壁碳納米管(CNTs)和Sc2O3雙促進的共沉淀型Pd-ZnO催化劑,其對MSR選擇生成CO2+H2顯示出優異的催化活性和操作穩定性.在組成經優化的CNTs單促進的Pd0.15Zn1-10%CNTs(10%表示質量分數,下同)催化劑上,在0.5 MPa,548 K,n(CH3OH)∶n(H2O)∶n(N2)=30∶30∶40和空速(GHSV)=180 L/(h·g)的反應條件下,反應150 h時H2的STY大體上維持在0.96 mol/(h·g)的高水平;這個值是非促進原基質Pd0.15Zn1催化劑的相應值(0.56 mol/(h·g))的1.7倍.摻入少量Sc2O3于上述CNTs單促進催化劑中能進一步提高其催化活性和操作穩定性.在組成經優化的雙促進的Pd0.15Zn1Sc0.067-10%CNTs催化劑上,在上述反應條件下,反應150 h時STY(H2)能穩定地保持在1.29 mol/(h·g)的更高水平;這個值是上述非促進的原基質Pd0.15Zn1催化劑的相應值的2.3倍(見圖1和表1).
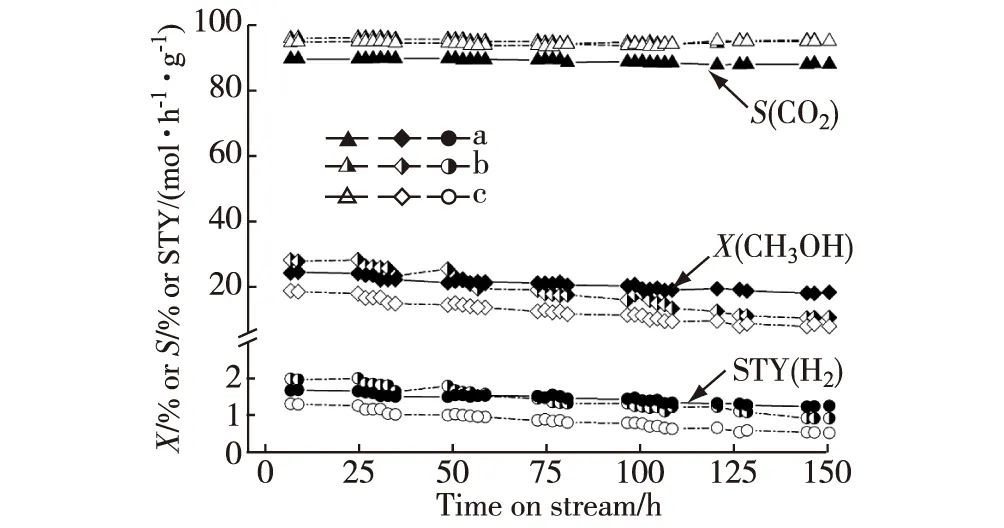
a.Pd0.15Zn1Sc0.067-10%CNTs;b.Pd0.15Zn1-10%CNTs;c.Pd0.15Zn1;反應條件:0.5 MPa,548 K,n(CH3OH)∶n(H2O)∶n(N2)=30∶30∶40,GHSV=180 L/(h·g).
上述MSR反應的表觀活化能(Ea) 的測試結果顯示,在Pd0.15Zn1Sc0.067-10%CNTs,Pd0.15Zn1-10%CNTs 和 Pd0.15Zn13種催化劑上觀測到的Ea值分別為86.2,86.4和87.8 kJ/mol.這些Ea值彼此相當接近,表明適當添加少量CNTs或CNTs+Sc2O3于Pd0.15Zn1基質催化劑中并不引起MSR反應的表觀活化能發生明顯變化,暗示少量CNTs或CNTs+Sc2O3的摻入并不導致MSR反應主要途徑的速率決定步驟有所改變.

(a) Pd0.15Zn1Sc0.067-10%CNTs;(b) Pd0.15Zn1-10%CNTs;(c) Pd0.15Zn1.
從反應后催化劑的透射電鏡(TEM)照片(圖2)可以估算,3種催化劑(Pd0.15Zn1Sc0.067-10%CNTs,Pd0.15Zn1-10%CNTs和Pd0.15Zn1)在MSR反應后試樣的PdZn合金微晶粒徑分別為4~7 nm,10~16 nm 和 12~16 nm.相應的X射線能譜(EDX)測量揭示,MSR反應后3種催化劑試樣的元素組成分別為:n(Pd)∶n(Zn)∶n(O)=6.4∶38.7∶54.9,n(Pd)∶n(Zn)∶n(O)∶n(C)=4.0∶28.9∶39.2∶27.9和n(Pd)∶n(Zn)∶n(O)∶n(C)∶n(Sc)=3.7∶24.6∶44.4∶26.7∶0.6.這表明:添加少量Sc2O3于Pd0.15Zn1-10%CNTs催化劑中導致工作態催化劑所含元素O的摩爾分數明顯上升,暗示還原為Pd0的Pd量占總Pd量的摩爾分數明顯下降.
XRD觀測結果(圖3)顯示,MSR反應后準工作態催化劑Pd和Zn組分可觀測到的主要晶相是ZnO和PdZn合金,單純金屬Pd相的含量在XRD觀測極限以下.利用Scherrer公式,可估算出3種催化劑(Pd0.15Zn1Sc0.067-10%CNTs,Pd0.15Zn1-10%CNTs和Pd0.15Zn1)的PdZn合金微晶粒徑分別為4.5,12.5和14.1 nm.這個結果與上述TEM照片(圖2)估算的結果十分相近,并表明隨著少量Sc3+的加入,PdZn合金的分散度有明顯提高.
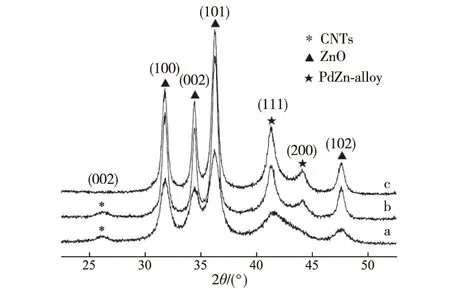
a.Pd0.15Zn1Sc0.067-10%CNTs;b.Pd0.15Zn1-10%CNTs;c.Pd0.15Zn1.
如圖4所示,X射線光電子能譜分析(XPS)揭示,3種MSR反應后準工作態催化劑的Pd(3d)-XPS譜都包含2或3種表面Pd物種:Pd0(PdZn合金),Pd2+(PdO)和Pd4+(PdO2).反應后的Pd0.15Zn1-10%CNTs催化劑的Pd0-物種占其總表面Pd的摩爾分數達到79.9%(見圖4-b和表2),高于非促進的基質Pd0.15Zn1催化劑的對應值(71.8%)(見圖4-c和表2),暗示適當量CNTs的加入改善了催化劑的可還原性,使表面Pd0-物種占總表面Pd的摩爾分數有所提高.這個結果連同上述XRD分析和催化劑活性評價的結果再次證實:以PdZn合金形式存在的表面Pd0-物種與相應催化劑對MSR制氫的優異選擇性存在密切相關性;高濃度的PdZn合金形式的表面Pd0-物種有助于MSR反應中H2和CO2的選擇生成.
與添加CNTs的情形不同,少量Sc3+的加入明顯地提高工作態催化劑表面Pd2+及Pd4+的摩爾分數;在反應后準工作態Pd0.15Zn1Sc0.067-10%CNTs催化劑表面,Pd2+和 Pd4+表面物種的摩爾分數上升至42.7%和14.4%,相應Pd0降為42.9%(見圖4-a和表2).Sc3+的顯著調變作用很可能緣于Sc2O3在ZnO晶格中的高溶解度,因為Sc3+的離子半徑(0.074 5 nm)與Zn2+(0.074 nm)十分相近.少量Sc2O3在ZnO晶格中的溶解導致陽離子形式的Schottky缺陷的生成,除非通過溶解入等價量的Pd+得以補償.然而,Pd+并不穩定,且由于其大得多的離子半徑使其無法溶解于ZnO晶格中(已知r(Pd2+)=0.085 nm,可期r(Pd+)更大).于是,由Sc2O3在ZnO晶格中的溶解而產生的陽離子形式Schottky缺陷將轉移/擴散到ZnO表面層,形成表面正離子空位;后者可通過裝填(PdZn)0-Pdn+原子簇的Pdn+(n=1或2)以達到對“摻雜有Sc3+的ZnO次表面層”的價態和電荷補償,(PdZn)0-Pdn+原子簇由此可得到穩定化.這將有助于抑制(PdZn)0納米顆粒的遷移和燒結,顯著延長催化劑的壽命.
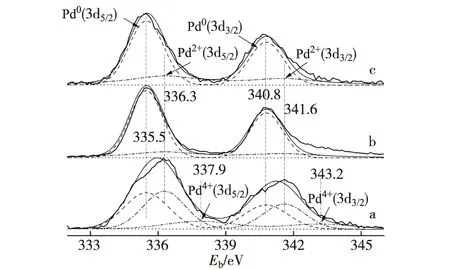
a.Pd0.15Zn1Sc0.067-10%CNTs;b.Pd0.15Zn1-10%CNTs;c.Pd0.15Zn1.
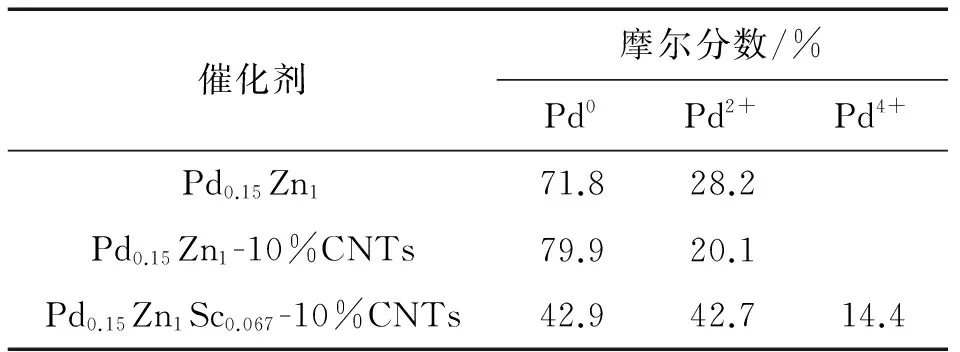
表2 反應后催化劑表面不同價態Pd物種的XPS結合能及相對含量
H2在預還原態催化劑上吸附的程序升溫脫附(H2-TPD)測量結果(圖5)顯示,在473~823 K溫度范圍,3種催化劑的H2-TPD曲線覆蓋區的相對面積強度比可估算為:S′(a)∶S′(b)∶S′(c)= 100∶56∶47,意味著3種催化劑工作態表面H2-吸附位濃度高低順序為:Pd0.15Zn1Sc0.067-10%CNTs > Pd0.15Zn1-10%CNTs > Pd0.15Zn1.基于CNTs高的電子傳遞性能及促進氫溢流的特性,在CNTs促進PdZn/ZnO催化的MSR反應過程中,從甲醇(及其中間產物HCHO和HCOOH)脫氫產生的 H(a) 物種,可以從PdZn/ZnO吸附位原位遷移至CNTs表面H-吸附位,隨后組合成H2(a),接著脫附為H2(g).這與Cu10Cr1/CNTs催化甲醇深度脫氫生成H2和CO的情景[28-29]相似,有助于提高MSR反應過程中一系列表面脫氫反應的速率.上述3種催化劑工作態表面H2-吸附位濃度增加順序與所觀測這些催化劑上MSR的反應活性高低順序相一致.
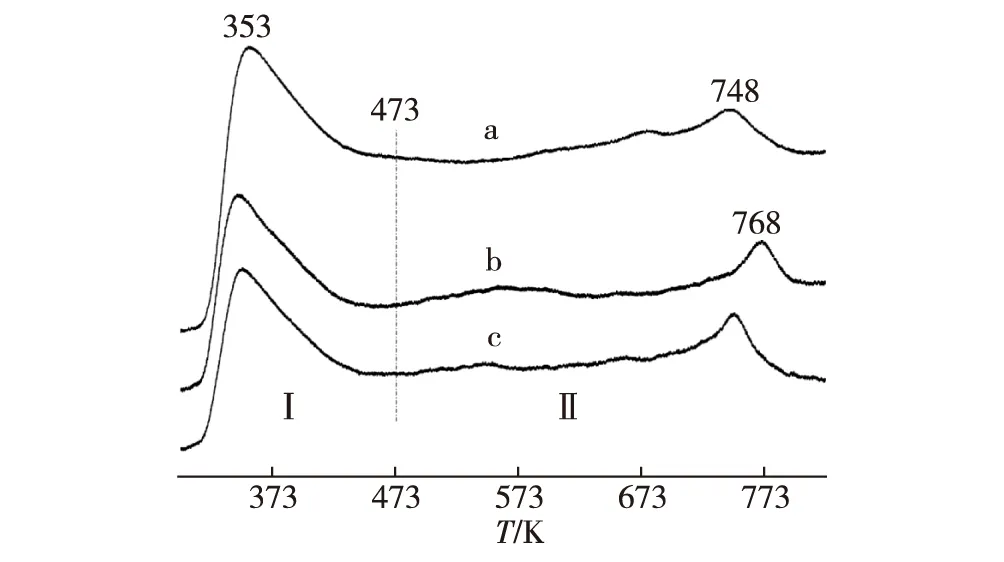
a.Pd0.15Zn1Sc0.067-10%CNTs;b.Pd0.15Zn1-10%CNTs;c.Pd0.15Zn1.
3 乙醇制氫研究概況
在理論上,乙醇和甲醇都可通過直接裂解、水蒸氣重整、部分氧化、或氧化重整等方法轉化產生H2,但若從可再生能源資源利用考慮,未來利用生物質經發酵法生產“生物乙醇”必將成為主流.生物質在成長過程中能吸收大量CO2,盡管乙醇生產、制氫須放出相當量的CO2,但整個過程形成一個碳循環,不凈產CO2排放;由此可期,乙醇制氫將是一條很具發展前景的制氫途徑.
現在乙醇制氫途徑主要有如下3條:1) 乙醇水蒸氣重整(ESR)制氫,2) 乙醇部分氧化制氫,3) 乙醇氧化重整制氫.
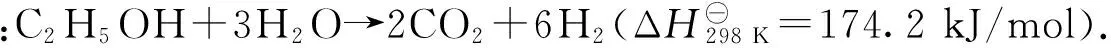
乙醇部分氧化制氫是利用燃料乙醇在氧氣不足的情況下發生氧化還原反應[34-35],反應式為:C2H5OH + 1.5O2→ 2CO2+ 3H2.該反應屬于放熱反應,不需外部供熱;其缺點是反應時需要純氧,成本較高,若用空氣進行反應,則產物中H2含量被稀釋,從而導致燃料電池效率下降,并增加凈化難度.
乙醇氧化重整制氫是將上述的“蒸氣重整”(吸熱)和“部分氧化”(放熱)兩個反應組合到一起(反應式為:C2H5OH + 2H2O+0.5O2→2CO2+ 5H2),并在一定條件下實現熱量自平衡[36-37];其優點是效率高、熱量得到充分利用,是一種最具發展潛力和工業應用價值的乙醇制氫途徑.
4 ESR制氫催化劑的研發
4.1 研發概況
在ESR制氫反應中,催化劑起著關鍵作用.高活性的催化劑應促使H2的生成達到最大化,同時能抑制結焦以及CH4的生成.盡管貴金屬的價格昂貴,但它具有負載型過渡金屬催化劑所不能代替的優良催化性能,因此負載型貴金屬催化劑的研究和開發仍是人們關注的熱點.現階段,貴金屬主要采用Rh,Ru,Pd,Pt等,使用的氧化物載體一般有Al2O3,MgO,ZrO2,CeO2,La2O3,TiO2等[35-44].
Liguras 等[38]比較了金屬負載量(即質量分數)在0~5%時的Al2O3(或MgO,TiO2)負載的 Rh,Ru,Pd和Pt催化劑在873~1 123 K溫度下的催化性能,以Rh催化劑上乙醇轉化率和H2的產率為最高.Ru催化劑在低負載量時幾無活性,但高負載量時則可與Rh相比;5%Ru/Al2O3能完全轉化乙醇為合成氣,生成H2的選擇性約95%.
載體的優化選擇十分重要.酸性的載體(諸如γ-Al2O3)易誘導乙醇脫水生成乙烯,這往往是結焦的根源;用K中和酸性載體或采用堿性載體(諸如γ-La2O3和MgO),脫水能被抑制.5%Ru/Al2O3在反應操作100 h之后,乙醇轉化率已下降15%;5%Rh/Al2O3在反應操作100 h之后,其活性也大為下降[39].Frusteri等[40]評價了MgO負載Rh,Pd,Ni和Co催化劑上ESR制氫的反應性能,結果顯示,Rh/MgO催化劑上乙醇轉化率和操作穩定性均最佳;由于MgO具堿性,Rh/MgO上結焦的速率很低,催化劑的失活主要是金屬顆粒燒結所致.Aupretre等[41]在Mg-Al基尖晶石氧化物載體上沉積Rh,與Rh/Al2O3相比,其堿性明顯提高,酸性明顯下降,其操作穩定性大為改善.
一直以來,人們都在尋求開發出低貴金屬含量的高效催化劑,雙金屬催化劑是其重要研發方向.第二種金屬的加入可通過表面修飾或形成合金的方式調變催化劑的行為,以產生良好的促進效應.Kugai等[35]研究了Rh-Ni/CeO2雙金屬體系,發現組成為1%Rh-5%Ni/CeO2的催化劑對ESR具有非常好的催化性能;但Ni的含量過高或過低都不利于H2的生成,在相近的反應條件下1%Rh/CeO2對ESR的催化活性比10%Ni/CeO2高近1倍.他們認為,Rh斷裂C—C鍵的能力比Ni強,同時還能有效地活化C—H鍵,在ESR反應中起主要作用;而Ni的功能主要是通過水煤氣變換反應(WGS)將CO轉化為CO2和H2.與上述Rh的情形不同,Pd和Zn在ZnO載體上共沉積能導致PdZn合金的生成,后者有助于脫氫反應和H2的生成[43].
表3示出若干ESR貴金屬催化劑[38-40,42,44].就ESR生產H2而言,Rh比其他貴金屬(諸如Pt,Pd和Au)更有效;在較高溫度和較大金屬負載量條件下,Ru的催化性能可與Rh相比.CeO2,MgO和La2O3適宜于作為ESR用Rh基催化劑的載體;而用Al2O3作為載體難免導致催化劑失活.從長期操作的穩定性考慮,MgO作為載體顯示最佳的性能;對于Rh,La2O3也可以是合適的載體.
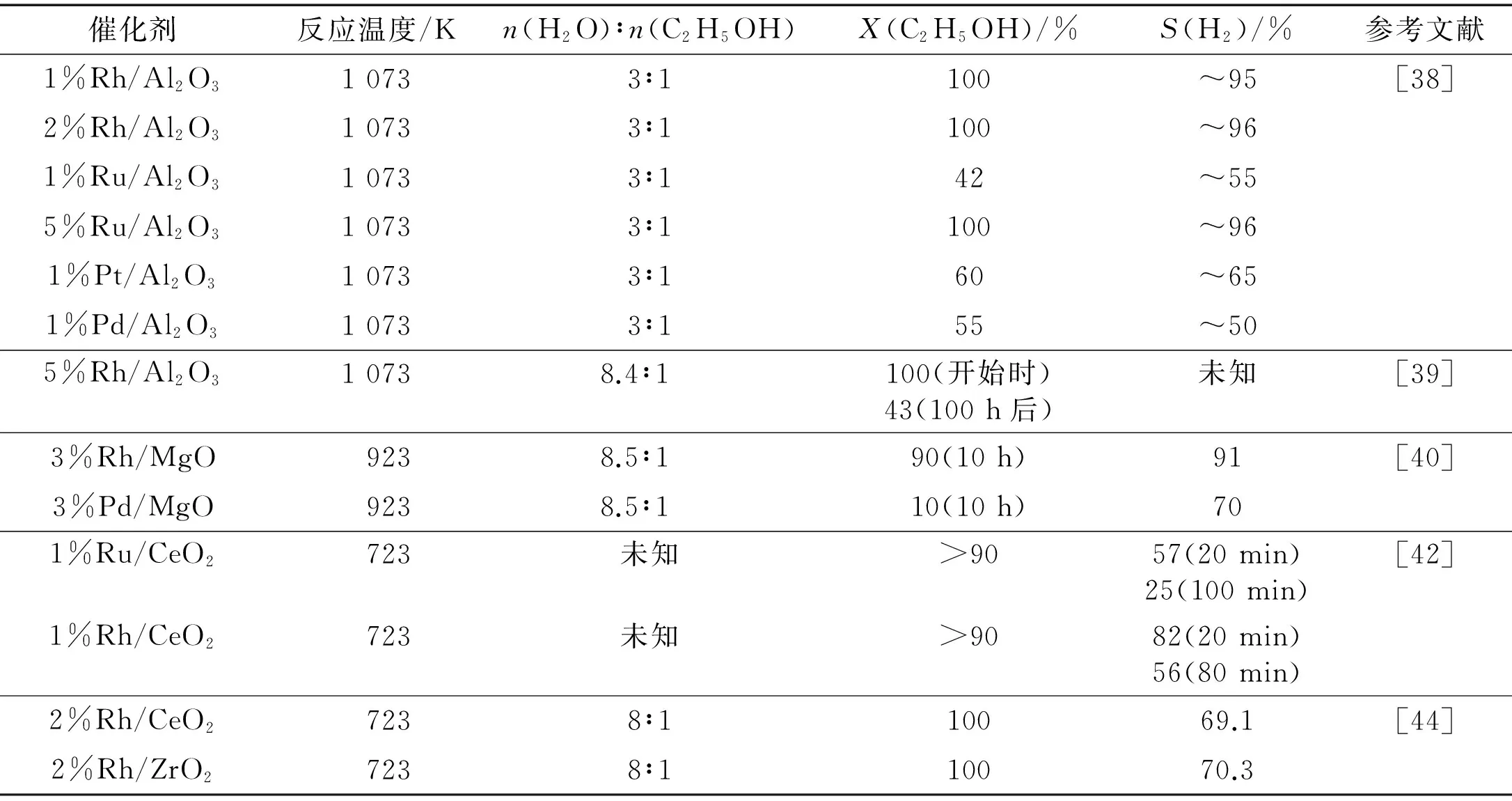
表3 貴金屬催化劑上ESR制H2的反應活性
由于金屬Rh的稀缺、昂貴,難有工業應用前景.非貴金屬ESR用作催化劑是著力研發的另一領域.迄今顯示出有興趣結果的催化劑主要是Ni基和Co基催化劑.Sun等[45]以草酸鎳作為前驅物,由浸漬-分解-還原法制備Ni/Y2O3,Ni/La2O3和Ni/Al2O33種催化劑,比較它們對ESR制氫的催化活性.在常壓,593 K的反應條件下,在Ni/Y2O3和Ni/La2O3上乙醇的轉化率分別可達93.1%和99.5%,H2的選擇性分別為53.2%和48.5%.Ni/La2O3催化劑的高活性和高穩定性是由于生成碳酸氧化鑭(La2O2CO3)物種,后者能與反應過程中沉積的表面碳反應,以防止催化劑失活.作為比較,Ni/Al2O3的H2選擇性在573 K達到最大值47.7%.上述報告的H2選擇性比較低很可能是由于所用反應原料氣的水/乙醇摩爾比較低(3∶1).Comas等[46]證實,增加水/乙醇摩爾比能顯著提高H2的選擇性.Yang等[47]評價Ni基催化劑上載體對ESR的影響,結果表明,在923 K和10%Ni負載量的條件下,參加試驗的4種催化劑上乙醇的轉化率幾乎都達到100%;H2選擇性的高低順序為:S(Ni/ZnO)≈S(Ni/La2O3)>S(Ni/MgO)>S(Ni/Al2O3).Frusteri等[48]評價在Ni/MgO催化劑上添加堿(Li或Na,K)對其催化性能的影響,結果顯示,添加Li和K能抑制Ni的燒結,從而提高催化劑的穩定性.他們[49]還發現,Ni/CeO2上積炭、結焦比Ni/MgO的快;這很可能與吸附在CeO2載體上的反應中間物種同載體之間發生強相互作用有關.
雙金屬或合金或復合氧化物負載的催化劑的研發興趣也有所增加.Barroso等[50]用檸檬酸鹽溶膠-凝膠法制備ESR制氫用的NiZnAl催化劑,發現產物分布對合金組成十分敏感.在Ni質量分數為18%~25%的催化劑上,在常壓、873 K、n(H2O)∶n(C2H5OH)=3.6~3.8的反應條件下,反應操作230 min,乙醇100%轉化,H2選擇性高達85%.Marino等[51]研究了Cu-Ni-K/γ-Al2O3對ESR制氫的催化活性,結果顯示:Cu和Ni分別促進脫氫和C—C鍵斷裂;K中和γ-Al2O3的酸性位有助于降低結焦的可能性.Bellido等[52]制備ZrO2,ZrO2-Y2O3和ZrO2-CaO負載的Ni催化劑,其用于催化ESR顯示,摻合Y2O3或CaO到ZrO2中能提高氣體產物中CO2和H2的含量,這種效應被歸因于載體中存在氧空位;當ZrO2用Y2O3修飾時H2選擇性上升,但用CaO修飾時反而下降,表明除氧空位之外,還有其他相互作用因素影響Ni物種的催化行為.Coleman等[53]評價一系列Mg-Al復合氧化物負載Ni(10%)催化劑對ESR制氫的催化性能,發現:復合氧化物負載Ni催化劑(10%Ni/(MgO-Al2O3))比單組分氧化物負載的對應物(10%Ni/MgO或10%Ni/Al2O3)具有較高的活性和生成H2和COx的選擇性;催化劑的性能與復合氧化物載體的Al和Mg含量存在相依性.他們認為,上述催化劑性能的改進與兩個單組分氧化物(MgO 和Al2O3)結合生成復合氧化物MgAl2O4相關;具有中等強度酸-堿性的MgAl2O4,通過降低副產物反應(生成乙烯和乙炔)的活性,從而提高催化劑催化ESR的活性、生成H2的選擇性以及操作穩定性.Akiyama等[54]制備ZrO2及其他載體負載、并經碳化的堿(或La,Ce)-摻雜的Ni催化劑,其催化ESR的結果顯示,經823 K碳化的Ni/ZrO2和Ni/V2O5展示了比CeO2,Al2O3,SiO2-Al2O3,和 MgO 負載的Ni催化劑更高的活性;與CeO2,Al2O3,SiO2-Al2O3,和 MgO 負載的Ni催化劑相比,摻雜Cs的Ni/ZrO2催化劑能更強烈地促進H2的生成.
已知負載的Co能斷裂C—C鍵[55],Co因而也是較廣泛研究的另一種非貴金屬催化劑.Haga等[56]較早報道:Co催化劑上乙醇催化轉化很大程度上受載體所控制.幾種載體負載的Co催化劑上ESR制H2的選擇性順序為:S(Co/Al2O3)>S(Co/ZrO2)>S(Co/MgO)>S(Co/SiO2)>S(Co/C).在Co/SiO2,Co/MgO和Co/ZrO2上可伴隨著CO加氫甲烷化,在Co/C上可伴隨著乙醇分解產生甲烷;而在Co/Al2O3上,CO甲烷化和乙醇分解兩個副反應都受到抑制,因此生成H2的選擇性最高.Cavallaro等[57]的工作顯示,由于MgO的堿性特征,在923 K下,Co/MgO比Co/Al2O3更能抑制焦炭的生成. Llorca等[58]用Co2(CO)8作為前驅物,制備ESR用的一種高度活潑Co/ZnO催化劑,在623 K下,其乙醇轉化率達100%,生成不含CO的H2的選擇性約為73%.75 h的試驗顯示其作為ESR活潑催化劑的操作穩定性和可實用性.他們的后續工作[59]顯示,添加Na能減少結焦,進一步提高Co/ZnO催化劑的活性和操作穩定性.Batista等[60]用浸漬法制備8.6%Co/Al2O3,7.8%Co/SiO2,和18%Co/MgO 3種催化劑,其催化ESR都顯示出高的活性(乙醇轉化率>90%) 和生成H2的選擇性(~70%).然而,在673 K反應9 h之后,3種催化劑都不同程度地結焦,其焦炭量順序為:Co/Al2O3(24.6%)>Co/MgO(17.0%)>Co/SiO2(14.2%).Al2O3上結焦最嚴重,這可歸因于Al2O3的酸性特性,有利于乙醇脫水生成乙烯.他們的后續研究[61]顯示,增加Co負載量能降低反應尾氣中的CO含量.Song等[62]研究γ-Al2O3,TiO2,ZrO2負載的Co催化劑,其催化ESR顯示,乙醇的轉化率同金屬分散度,也即金屬Co活性位,密切相關;產物分布取決于主反應與副反應(包括:ESR,甲烷化,WGS,脫水和脫氫)的競爭.在所用的幾種載體中,ZrO2能提供最高的金屬分散度和最高的H2產率:在823 K下,在10%Co/ZrO2上H2的產率達92%(即1 mol乙醇可以產出5.5 mol H2).Chiou等[63]用浸漬法制備Cox/ZnO和PtyCo10/ZnO催化劑,評價其對ESR的催化性能.組成經優化的10%Co/ZnO顯示出最高的表面積和最高的催化活性.(0.5%Pt-10%Co)/ZnO顯示出最佳的混合和協同效應,組合Pt和Co單金屬催化劑的優點,即在≤ 300 ℃的溫度下,Pt善于催化乙醇脫氫和乙醛分解,而Co善于催化乙醛重整.添加適量Pt于10%Co/ZnO中能提高ESR制氫的效率;在598 K下,在組成經優化的(0.5%Pt-10%Co)/ZnO催化劑上,產生最佳的結果,反應產物的選擇性為:n(H2)∶n(CO2)∶n(CH4)∶n(CO)≈73.2∶23.8∶2.4∶0.6.
4.2 高效高穩定性催化劑研發
新近本實驗室Hou等[64]研發出一種共沉淀型Yb2O3摻雜Ni-ZrO2催化劑.該催化劑對ESR選擇生成CO2和H2顯示出異常高的活性和熱穩定性.在組成經優化的Ni1.25Zr1Yb0.8催化劑上,在0.5 MPa,723 K,n(C2H5OH)∶n(H2O)∶n(N2)=12.5∶37.5∶50和GHSV=90 L/(h·g)的反應條件下,反應120 h時,乙醇轉化率保持在18.2%水平,相應的H2的STY為0.396 mol/(h·g).這個H2的STY值是非促進的原基質Ni1.25Zr1催化劑的相應值(0.247 mol/(h·g))的1.6倍(見圖6和表4).在上述反應條件下,歷時240 h的整個反應過程中,Ni1.25Zr1Yb0.8催化劑上的乙醇轉化率、CO2選擇性和H2的STY均能大體保持穩定.相比之下,不含Yb2O3的原基質Ni1.25Zr1催化劑上的乙醇轉化率、CO2選擇性和H2的STY均緩慢下降,反應240 h時已分別降至7%,65.1%和0.204 mol/(h·g)(見圖6).
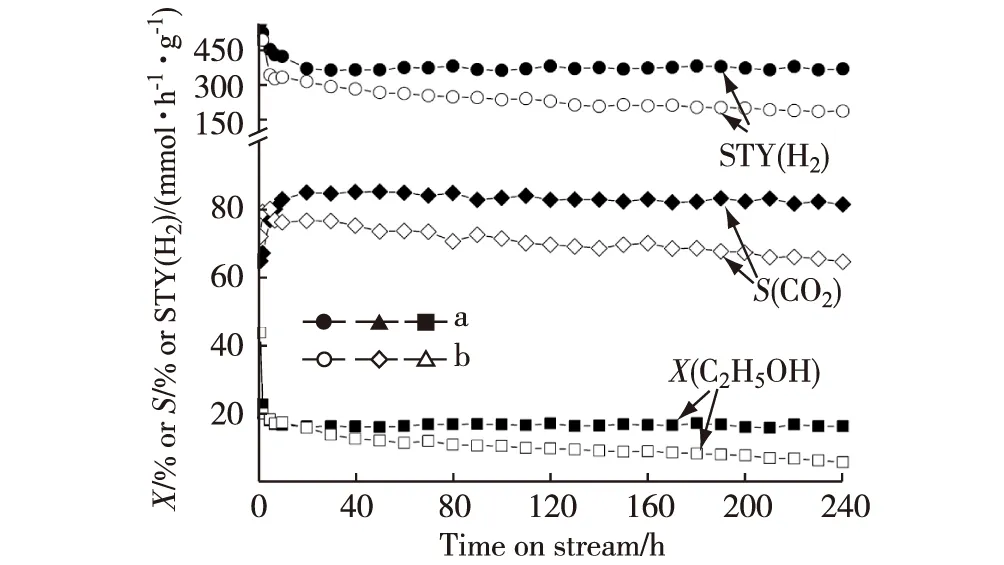
a.Ni1.25Zr1Yb0.8;b.Ni1.25Zr1;反應條件:0.5 MPa,723 K,n(C2H5OH)∶n(H2O)∶n(N2)=12.5∶37.5∶50,GHSV=90 L/(h·g).
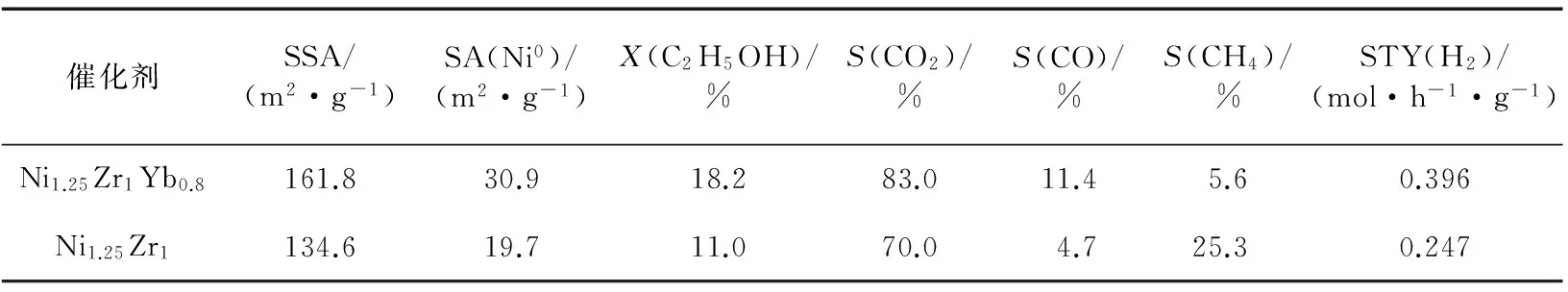
表4 在Yb2O3促進的Ni-ZrO2催化劑及其參比體系上ESR制氫的反應活性[64]
催化劑的耐熱性能優劣是其是否具備實用性的一項重要指標.所研發Ni1.25Zr1Yb0.8催化劑用于ESR制氫的耐熱性能測試在0.5 MPa,723~973 K,n(C2H5OH)∶n(H2O)∶n(N2)=12.5∶37.5∶50,GHSV=90 L/(h·g)的反應條件下進行,并與非促進的原基質Ni1.25Zr1催化劑作比較.在歷時180 h的耐熱試驗中,被測催化劑連續在723,823,873,923和973 K每個溫度點各工作24 h后將操作溫度降回至723 K進行活性測試,示于圖7的結果表明,Ni1.25Zr1Yb0.8催化劑上進行ESR反應的乙醇轉化率一直保持在19%以上,沒觀察到失活的跡象.而不添加Yb2O3的原基質Ni1.25Zr1催化劑差別懸殊,在823 K經24 h熱處理操作之后,其乙醇轉化率已降至4.4%;這種轉化率的突然下降意味著催化劑已燒結失活.

(a) Ni1.25Zr1Yb0.8;(b) Ni1.25Zr1.
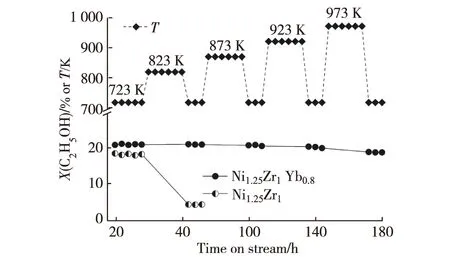
圖7 不同反應溫度下ESR制氫催化劑的熱穩定性[64]
上述催化劑上ESR反應表觀活化能的測試在0.5 MPa,673~773 K,n(C2H5OH)∶n(H2O)∶n(N2)=12.5∶37.5∶50,GHSV=105 L/(h·g)的反應條件下進行,結果顯示,Ni1.25Zr1Yb0.8和Ni1.25Zr12種催化劑上所觀測Ea值分別為74.5 和71.5 kJ/mol,彼此相當接近,表明適當添加Yb2O3于Ni1.25Zr1基質催化劑中并不引起ESR反應的表觀活化能發生明顯變化,暗示Yb2O3的參與并不導致ESR反應主要途徑的速率決定步驟有所改變.
圖8所示掃描電鏡(SEM)照片可見,反應240 h后準工作態催化劑Ni1.25Zr1Yb0.8和Ni1.25Zr1的外貌有明顯差異.在添加Yb2O3的催化劑上,Nix0-NiOy納米顆粒保持著相同的形狀和顆粒大小,且均勻分散在(Zr-Yb)Oz復合載體上.由于在SEM圖中很難將Nix0-NiOy納米顆粒和(Zr-Yb)Oz復合載體區別開來,遂將Nix0-NiOy/(Zr-Yb)Oz作為一個復合整體對待.經估算,Ni1.25Zr1Yb0.8催化劑上的Nix0-NiOy/(Zr-Yb)Oz顆粒粒徑約在15~30 nm;而Ni1.25Zr1催化劑上的Nix0-NiOy/ZrO2顆粒粒徑卻在60~100 nm,并在催化劑表面生展出可觀量的CNTs或碳納米纖維(CNFs).上述結果與2種催化劑工作態表面金屬Ni的表面積以及2種催化劑上ESR反應活性的高低順序相一致.
ESR反應240 h后催化劑的XRD測量結果(圖9)顯示,在添加Yb2O3的催化劑上觀測到的微晶相是Nix0和c-(Zr-Yb)Oy,其平均粒徑分別為4.5和4.8 nm;而不含Yb2O3的原基質催化劑上觀測到的微晶相是Nix0,t-ZrO2和m-ZrO2,其平均粒徑分別為15.6,12.6和14.6 nm.這表明不含Yb2O3的原基質催化劑,其Nix0和t-ZrO2微晶的粒徑隨著反應的進行明顯增大,并且有部分t-ZrO2轉變為更為穩定的m-ZrO2.基于上述結果可作如下論斷,由于Yb3+的離子半徑(0.087 nm)與Zr4+的(0.079 nm)較為接近,可以較大量地溶于ZrO2晶格中,從而形成具有高度熱穩定性的“立方氧化鋯結構”的Yb-Zr復合氧化物(c-(Zr-Yb)Oy)[65-68],這是Ni1.25Zr1Yb0.8催化劑展示出更高活性和熱穩定性的主要原因.
a.Ni1.25Zr1Yb0.8;b.Ni1.25Zr1.
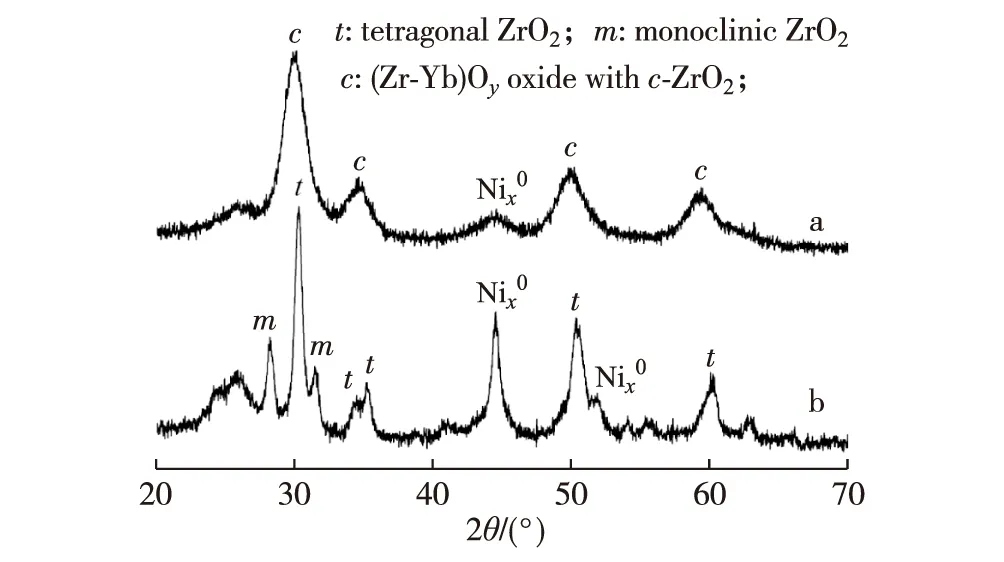
圖9 反應后催化劑Ni1.25Zr1Yb0.8(a)和Ni1.25Zr1(b)的XRD譜[64]
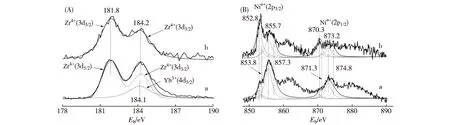
反應后(即準工作態)催化劑的XPS譜(圖10)揭示,添加Yb3+與不添加Yb3+的2種催化劑,它們的Zr(3d)-XPS譜的位置和形狀幾乎無變化,但Ni(2p)-XPS譜的形狀有明顯差別.在Ni1.25Zr1Yb0.8催化劑表面,其Ni物種的主要存在形態是Ni(OH)2,其次是Ni2O3,Ni0和NiO含量很少.而在Ni1.25Zr1催化劑表面,主要的Ni物種是Ni0,其次是Ni(OH)2,而Ni2O3和NiO含量甚低.那些不同價態或微環境的Ni物種的XPS結合能及摩爾分數示于表5.可以推斷,在準工作態的Ni1.25Zr1Yb0.8催化劑表面,Ni組分主要是以Nix0-Nin+(n= 2或3)的形式存在,這很可能是由于催化劑表面高度分散的Ni納米顆粒與c-(Zr-Yb)Oy載體之間存在較強的相互作用所致;通過其Nin+與表面O2-的結合,使Nix0-Nin+納米簇錨定在c-(Zr-Yb)Oy載體表面,避免活性組分Nix0納米顆粒的移動和聚集,保持表面金屬Ni的高分散度,有效抑制表面積碳,從而明顯地提高催化劑的熱穩定性和操作穩定性.
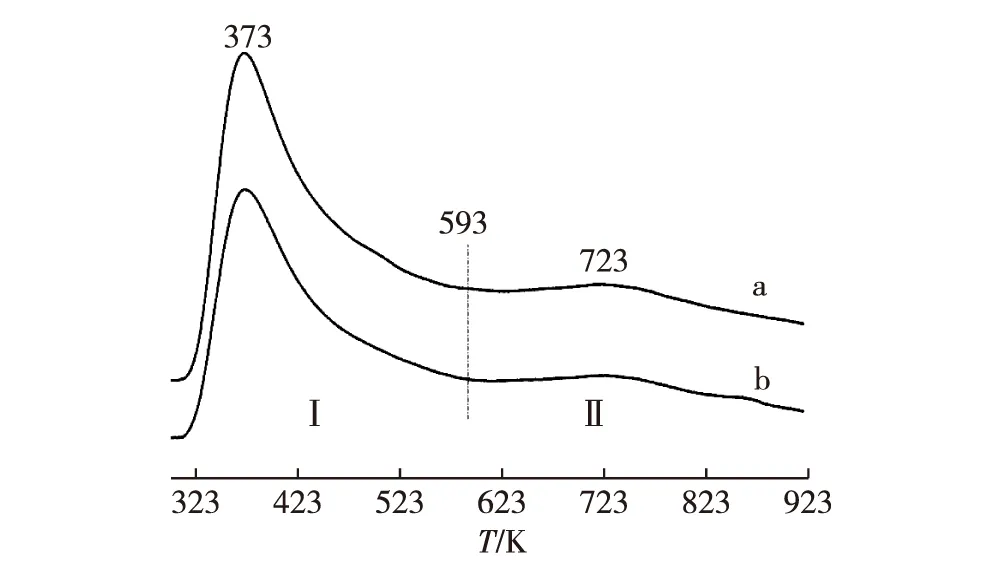
H2-TPD研究可為催化劑表面氫吸附位的濃度和性質提供有用信息.圖11示出2種催化劑的H2-TPD測試結果.可以推斷,每一條H2-TPD曲線的低溫段(303~593 K)系源于弱吸附氫物種(包括分子態吸附氫H2(a)和原子態弱吸附氫H(a))的脫附),而其高溫段(593~923 K)可歸因于原子態強吸附氫H(a)的脫附.可以想象,在ESR反應溫度(本工作≥593 K)下,與低溫段(即區-I)相關的吸附氫的表面濃度必然很低,在工作態催化劑表面大部分吸附氫是對應于高溫段(即區-II)的那些吸附氫.這暗示:正是那些較強的氫吸附位與ESR反應活性密切相關.在Ni-ZrO2基催化劑催化的ESR反應過程中,從反應物C2H5OH(以及中間產物CH3CHO和CH3COCH3)脫氫產生的氫吸附物種H(a)可以原位轉移到工作態催化劑表面的那些較強的H-吸附位,結合成H2(a),而后脫附成H2(g).這將有助于提高ESR反應過程中一系列表面脫氫反應的速率.被測試的2種催化劑在區-II的H2-TPD曲線覆蓋區的相對面積強度比可估算為S′(a)∶S′(b)=100∶60,意味著2種催化劑活性表面氫吸附位濃度的高低順序為:Ni1.25Zr1Yb0.8> Ni1.25Zr1,與所觀測2種催化劑上ESR反應活性的高低順序相一致.
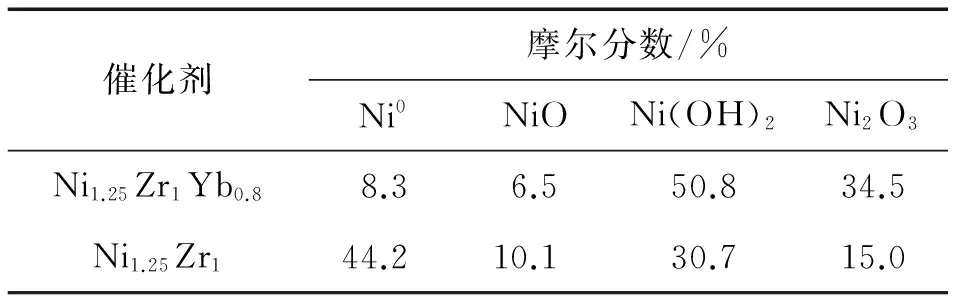
表5 反應后催化劑表面不同價態或微環境的Ni物種的XPS結合能及相對含量[64]
a.Ni1.25Zr1Yb0.8;b.Ni1.25Zr1.
5 結 語
在過去十多年,有關MSR和ESR制氫的研究已取得重要進展.雖然不同研究組之間仍存在一些引人注意的微妙差異,但有關MSR和ESR的反應機理以及催化劑的組成/結構,其認識已趨于一致,同時涌現出若干系列催化劑.
若從工業實用角度考慮,催化劑的操作穩定性(以及耐熱性和壽命)是一個挑戰性問題.催化劑失活主要緣于如下2個因素:1) 催化活性金屬顆粒的團聚燒結;2) 碳沉積、結焦;這些是遲早須研究解決的問題.現已知道,載體的組成/結構/性能對催化劑的操作穩定性和耐熱性能有重大影響:載體的酸堿性影響著主反應與副反應(包括:ESR,甲烷化,WGS,脫水和脫氫)的競爭,由此影響產物的選擇性;載體自身是否不易燒結(即具有高分散性及其耐熱性)、并具有穩定化高度分散載于其表面的金屬活性組分的性能,這些屬性與結焦和金屬活性微晶的燒結密切相關.上述本研究組研發的2種高活性、高穩定性的催化劑,想必可為工業實用型MSR和ESR催化劑的開發提供一些有益思路.
[1] 陳丹之.氫能[M].西安:西安交通大學出版社,1990.
[2] 葉大均.能源概論[M].北京:清華大學出版社,1990.
[3] Dicks A L.Hydrogen generation from natural gas for the fuel cell systems of tomorrow [J].J Power Sources,1996,61:113-124.
[4] Mitsugi C,Harumi A,Kenzo F.We-net:Japanese hydrogen program [J].Int J Hydrogen Energy,1998,23:159-165.
[5] Das L M.On-board hydrogen storage systems for automotive application [J].Int J Hydrogen Energy,1996,21:789-800.
[6] 陳進富,趙永豐,朱亞杰.儲氫技術及其發展現狀 [J].化工進展,1997(1):10-15.
[7] Donitz W.Fuel cells for mobile applications,status,requirements and future application potential [J].Int J Hydrogen Energy,1998,23:611-615.
[8] Thomas C E,Kuhn I F,James B D,et al.Affordable hydrogen supply pathways for fuel cell vehicles [J].Int J Hydrogen Energy,1998,23:507-516.
[9] Cannon J S.Hydrogen vehicle programs in the USA [J].Int J Hydrogen Energy,1994,19:905-909.
[10] Amphlett J C,Creber K A M,Davis J M,et al.Hydrogen-production by steam reforming of methanol for polymer electrolyte fuel-cells [J].Int J Hydrogen Energy,1994,19:131-137.
[11] Hohlein B,Boe M,Bogild Hansen J,et al.Hydrogen from methanol for fuel cells in mobile systems:development of a compact reformer [J].J Power Sources,1996,61:143-147.
[12] Cheng W H.Development of methanol decomposition catalysts for production of H2and CO [J].Acc Chem Res,1999,32:685-691.
[13] Gottesfeld S,Pafford J.A new approach to the problem of carbon-monoxide poisoning in fuel-cells operating at low-temperatures [J].J Electrochem Soc,1988,135:2651-2652.
[14] Amphlett J C,Mann R F,Peppley B A.On board hydrogen purification for steam reformation PEM fuel cell vehicle power plants [J].Int J Hydrogen Energy,1996,21:673-678.
[15] Trimm D L,?nsan Z I.On-board fuel conversion for hydrogen-fuel-cell-driven vehicles [J].Catal Rev Sci Eng,2001,43:31-84.
[16] Tsai A P,Yoshimura M.Highly active quasi-crystalline Al-Cu-Fe catalyst for steam reforming of methanol [J].Appl Catal A:Gen,2001,214:237-241.
[17] Liu Y,Hayakawa T,Suzuki K,et al.Highly active copper/ceria catalysts for steam reforming of methanol [J].Appl Catal A:Gen,2002,223:137-145.
[18] Zhang X R,Shi P,Zhao J,et al.Production of hydrogen for fuel cells by steam reforming of methanol on Cu/ZrO2/Al2O3catalysts [J].Fuel Process Technol,2003,83:183-192.
[19] Iwasa N,Kudo S,Takahashi H,et al.Highly selective supported Pd catalysts for steam reforming of methanol [J].Catal Lett,1993,19:211-216.
[20] Iwasa N,Masuda S,Ogawa N,et al.Steam reforming of methanol over Pd/ZnO:effect of the formation of PdZn alloys upon the reaction[J].Appl Catal A:Gen,1995,125:145-157.
[21] Iwasa N,Mayanagi T,Ogawa N,et al.New catalytic functions of Pd-Zn,Pd-Ga,Pd-In,Pt-Zn,Pt-Ga and Pt-In alloys in the conversions of methanol [J].Catal Lett,1998,54:119-123.
[22] Iwasa N,Mayanagi T,Nomura W,et al.Effect of Zn addition to supported Pd catalysts in the steam reforming of methanol [J].Appl Catal A:Gen,2003,248:153-160.
[23] Karim A,Conant T,Datye A.The role of PdZn alloy formation and particle size on the selectivity for steam reforming of methanol [J].J Catal,2006,243:420-427.
[24] Chin Y H,Wang Y,Dagle R A,et al.Methanol steam reforming over Pd/ZnO:catalyst preparation and pretreatment studies [J].Fuel Process Technol,2003,83:193-201.
[25] Lorenz H,Penner S,Jochum W,et al.Pd/Ga2O3methanol steam reforming catalysts:Part II.Catalytic selectivity [J].Appl Catal A:Gen,2009,358:203-210.
[26] Halevi B,Peterson E J,Roy A,et al.Catalytic reactivity of face centered cubic PdZn α for the steam reforming of methanol [J].J Catal,2012,291:44-54.
[27] Yang L,Lin G D,Zhang H B.Highly efficient Pd-ZnO catalyst doubly promoted by CNTs and Sc2O3for methanol steam reforming [J].Appl Catal A:Gen,2013,455:137-144.
[28] 陳書貴.周金梅,張鴻斌,等.碳納米管負載/促進Cu-Cr催化劑上甲醇分解制氫[J].廈門大學學報:自然科學版,2003,42(2):133-138.
[29] Zhang H B,Dong X,Yuan Y Z,et al.MWCNTs as novel material for carrier or promoter of catalyst[C]//13thICC.Paris,France:[s.n.],2004:267.
[30] Mattos L V,Noronha F B.Partial oxidation of ethanol on supported Pt catalysts[J].J Power Sources,2005,145:10-15.
[31] Bi L,Hsu S N,Yeh C T,et al.Low-temperature mild partial oxidation of ethanol over supported platinum catalysts [J].Catal Today,2007,129:330-335.
[32] Fierro V,Akidim O,Mirodatos C.On-board hydrogen production in a hybrid electric vehicle by bio-ethanol oxidative steam reforming over Ni and noble metal based catalysts [J].Green Chem,2003,5:20-24.
[33] Pereira E B,Homs N,Marti S,et al.Oxidative steam-reforming of ethanol over Co/SiO2,Co-Rh/SiO2and Co-Ru/SiO2catalysts:catalytic behavior and deactivation/ regeneration processes [J].J Catal,2008,257:206-214.
[34] Auprêtre F,Descorme C,Duprez D.Bio-ethanol catalytic steam reforming over supported metal catalysts [J].Catal Commun,2002,3:263-267.
[35] Kugai J,Subramani V,Song C,et al.Effects of nanocrystalline CeO2supports on the properties and performance of Ni-Rh bimetallic catalyst for oxidative steam reforming of ethanol [J].J Catal,2006,238:430-440.
[36] Cai W,Wang F,Zhan E,et al.Hydrogen production from ethanol over Ir/CeO2catalysts:a comparative study of steam reforming,partial oxidation and oxidative steam reforming [J].J Catal,2008,257:96-107.
[37] de Lima S M,Silva A M,Graham U M,et al.Ethanol decomposition and steam reforming of ethanol over CeZrO2and Pt/CeZrO2catalyst:reaction mechanism and deactivation [J].Appl Catal A:Gen,2009,352:95-113.
[38] Liguras D K,Kondarides D I,Verykios X E.Production of hydrogen for fuel cells by steam reforming of ethanol over supported noble metal catalysts [J].Appl Catal B:Env,2003,43:345-354.
[39] Cavallaro S,Chiodo V,Freni S,et al.Performance of Rh/Al2O3catalyst in the steam reforming of ethanol:H2production for MCFC [J].Appl Catal A:Gen,2003,249:119-128.
[40] Frusteri F,Freni S,Spadaro L,et al.H2production for MC fuel cell by steam reforming of ethanol over MgO supported Pd,Rh,Ni and Co catalysts [J].Catal Commun,2004,5:611-615.
[41] Aupretre F,Descorme C,Duprez D,et al.Ethanol steam reforming over MgxNi1-xAl2O3spinel oxide-supported Rh catalysts [J].J Catal,2005,233:464-477.
[42] Erdohelyi A,Rasko J,Kecskes T,et al.Hydrogen formation in ethanol reforming on supported noble metal catalysts [J].Catal Today,2006,116:367-376.
[43] Casanovas A,Llorca J,Homs N,et al.Ethanol reforming processes over ZnO-supported palladium catalysts:effect of alloy formation [J].J Mol Catal A:Chem,2006,250:44-49.
[44] Diagne C,Idriss H,Pearson K,et al.Efficient hydrogen production by ethanol reforming over Rh catalysts effect of addition of Zr on CeO2for the oxidation of CO to CO2[J].C R Chim,2004,7:617-622.
[45] Sun J,Qiu X P,Wu F,et al.H2from steam reforming of ethanol at low temperature over Ni/Y2O3,Ni/La2O3and Ni/Al2O3catalysts for fuel-cell application [J].Int J Hydrogen Energy,2005,30:437-445.
[46] Comas J,Marino F,Laborde M,et al.Bio-ethanol steam reforming on Ni/Al2O3catalyst [J].Chem Eng J,2004,98:61-68.
[47] Yang Y,Ma J X,Wu F.Production of hydrogen by steam reforming of ethanol over a Ni/ZnO catalyst [J].Int J Hydrogen Energy,2006,31:877-882.
[48] Frusteri F,Freni S,Chiodo V,et al.Steam reforming of bio-ethanol on alkali-doped Ni/MgO catalysts:hydrogen production for MC fuel cell [J].Appl Catal A:Gen,2004,270:1-7.
[49] Frusteri F,Freni S,Chiodo V,et al.Steam and auto-thermal reforming of bio-ethanol over MgO and CeO2Ni supported catalysts [J].Int J Hydrogen Energy,2006,31:2193-2199.
[50] Barroso M N,Gomez M F,Arrua L A,et al.Hydrogen production by ethanol reforming over NiZnAl catalysts [J].Appl Catal A:Gen,2006,304:116-123.
[51] Marino F,Baronetti G,Jobbagy M,et al.Cu-Ni-K/γ-Al2O3supported catalysts for ethanol steam reforming formation of hydrotalcite-type compounds as a result of metal-support interaction [J].Appl Catal A:Gen,2003,238:41-54.
[52] Bellido J D A,Assaf E M.Nickel catalysts supported on ZrO2,Y2O3-stabilized ZrO2and CaO-stabilized ZrO2for the steam reforming of ethanol:effect of the support and nickel load [J].J Power Sources,2008,177:24-32.
[53] Coleman L J I,Epling W,Hudgins R R,et al.Ni/Mg-Al mixed oxide catalyst for the steam reforming of ethanol [J].Appl Catal A:Gen,2009,363:52-63.
[54] Akiyama M,Oki Y,Nagai M.Steam reforming of ethanol over carburized alkali-doped nickel on zirconia and various supports for hydrogen production [J].Catal Today,2012,181:4-13.
[55] Llorca J,Homs N,Piscina P R.In situ DRIFT-mass spectrometry study of the ethanol steam-reforming reaction over carbonyl-derived Co/ZnO atalysts [J].J Catal,2004,227:556-560.
[56] Haga F,Nakajima T,Miya H,et al.Catalytic properties of supported cobalt catalysts for steam reforming of ethanol [J].Catal Lett,1997,48:223-227.
[57] Cavallaro S,Mondello N,Freni S.Hydrogen produced from ethanol for internal reforming molten carbonate fuel cell [J].J Power Sources,2001,102:198-204.
[58] Llorca J,Piscina P R,Dalmon J A,et al.CO-free hydrogen from steam reforming of bio-ethanol over ZnO-supported cobalt catalysts:effect of the metallic precursor [J].Appl Catal B:Env,2003,43:355-369.
[59] Llorca J,Homs N,Sales J,et al.Effect of sodium addition on the performance of Co-ZnO-based catalysts for hydrogen production from bio-ethanol [J].J Catal,2004,222:470-480.
[60] Batista M S,Santos R K S,Assaf E M,et al.Characterization of the activity and stability of supported cobalt catalysts for the steam reforming of ethanol[J].J Power Sources,2003,124:99-103.
[61] Batista M S,Santos R K S,Assaf E M,et al.High efficiency steam reforming of ethanol by cobalt-based catalysts [J].J Power Sources,2004,134:27-32.
[62] Song H,Zhang L,Watson R B,et al.Investigation of bio-ethanol steam reforming over cobalt-based catalysts [J].Catal Today,2007,129:346-354.
[63] Chiou J Y Z,Wang W Y,Yang S Y,et al.Ethanol steam reforming to produce hydrogen over Co/ZnO and PtCo/ZnO catalysts [J].Catal Lett,2013,143:501-507.
[64] Hou J,Liu Z M,Lin G D,et al.Novel Ni-ZrO2catalyst doped with Yb2O3for ESR [J].Int J Hydrogen Energy,2014,39:1315-1324.
[65] Martin U,Boysen H,Frey F.Neutron powder investigation of tetragonal and cubic stabilized zirconia,TZP and CSZ,at temperatures up to 1 400 K[J].Acta Cryst,1993,B49:403-413.
[66] Hypp?nen I,H?ls? J,Kankare J,et al.Defect structure and up-conversion luminescence properties of ZrO2:Yb3+,Er3+nanomaterials [J].J Fluoresc,2008,18:1029-1034.
[67] Angeles-Chavez C,Salas P,Díaz-Torres L A,et al.Structural and chemical characterization of Yb2O3-ZrO2system by HAADF-STEM and HRTEM [J].Microsc Microanal,2009,15:46-53.
[68] Solis D,de la Rosa E,Meza O,et al.Role of Yb3+and Er3+concentration on the tunability of green-yellow-red upconversion emission of codoped ZrO2:Yb3+-Er3+nanocrystals [J].J Appl Phys,2010,108:023103.
Development of Novel Catalysts for Steam-reforming of Methanol or Ethanol to Generate Hydrogen
LIANG Xue-lian,LIU Zhi-ming,XIE Jian-rong,CHEN Bing-hui,LIN Guo-dong,ZHANG Hong-bin*
(State Key Laboratory of Physical Chemistry of Solid Surfaces,National Engineering Laboratory for Green Chemical Productions of Alcohols-Ethers-Esters,College of Chemistry and Chemical Engineering,Xiamen University,Xiamen 361005,China)
There is an ongoing trend to move toward exploitation of clean alternative energy sources.As one of the ideal energy sources,hydrogen has drawn great attention and been intensively studied.In order for hydrogen to be more widely used,especially serving as power-fuel for hydrogen-fuel-cell-driven vehicles,it is critical to solve issues associated with the storage and transportation of hydrogen.Liquid hydrogen-carriers,which can generate hydrogen in situ through conversion/reforming,are commercially important for practical applications.Among the many liquid-fuels that can generate hydrogen through reforming,methanol and ethanol have displayed some advantages in relatively lower reaction temperature and pressure,higher H/C ratio,no emission of NOxand SOxas by-products,and making use of the existing power-fuel transportation-distribution systems.In this article,we review recent developments in the hydrogen generation from methanol or ethanol,highlighting the progress in our lab on the development of catalysts for hydrogen generation from methanol or ethanol through steam reforming.
hydrogen energy;methanol steam-reforming;generate hydrogen;ethanol steam-reforming
2015-03-24 錄用日期:2015-06-10
國家重點基礎研究發展計劃(973計劃)(2011CBA00508);優秀國家重點實驗室基金項目(20923004);教育部創新團隊項目(IRT1036)
梁雪蓮,劉志銘,謝建榕,等.甲醇或乙醇水蒸氣重整制氫高效新型催化劑的研發[J].廈門大學學報:自然科學版,2015,54(5):693-706.
:Liang Xuelian,Liu Zhiming,Xie Jianrong,et al.Development of novel catalysts for steam-reforming of methanol or ethanol to generate hydrogen[J].Journal of Xiamen University:Natural Science,2015,54(5):693-706.(in Chinese)
10.6043/j.issn.0438-0479.2015.05.013
新能源材料專題
O 643.36
A
0438-0479(2015)05-0693-14
* 通信作者:hbzhang@xmu.edu.cn
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
