擬南芥蔗糖合酶基因合成、重組表達及其在酶催化合成萊鮑迪甙A中的初步應用
2015-09-04 10:23:02陳量量成采虹陳可泉
食品工業(yè)科技 2015年23期
杜 婷,王 鈺,陳量量,成采虹,李 艷,陳可泉
(南京工業(yè)大學生物與制藥工程學院,江蘇南京211816)
蔗糖合酶(EC2.4.1.13,sucrose synthase,SuSy)是植物蔗糖代謝的關鍵酶之一。該酶可催化蔗糖和UDP(二磷酸尿核苷)生成果糖和UDPG(尿苷二磷酸葡萄糖)的可逆反應,但目前研究認為該酶偏向于蔗糖裂解反應方向[1-3]。SuSy的活性可被己糖抑制并造成裂解反應的中止[4]。反應生成的UDPG是一種重要的尿苷二磷酸單糖,可在UDP糖基轉移酶所催化的葡萄糖基轉移反應中作為輔底物提供活化的葡萄糖基[5]。利用蔗糖合酶與糖基轉移酶的偶聯可使反應體系中的UDPG循環(huán)再生[6-8]。實驗室前期研究的甜葉菊糖基轉移酶UGT76G1能特異性催化甜菊糖中含量高并且具有較強后苦味的ST甙(甜菊糖甙)生成高甜度的RA甙(萊鮑迪甙A)[9],其催化反應由UDPG提供葡萄糖基。采用全細胞催化技術,通過外加葡萄糖生成胞內UDPG,可避免添加昂貴的UDPG作為反應底物[10],但涉及ST甙和RA甙進出細胞的問題,影響了催化效率。本研究中采用基于蔗糖合酶再生UDPG的雙酶體系催化ST甙合成RA甙(圖1)。先采用兩步PCR合成蔗糖合酶AtSUS1的編碼基因,然后構建其與糖基轉移酶UGT76G1共表達的重組菌。采用重組菌制備粗酶液進行催化反應,并考察了UDP起始濃度、pH、溫度,以及反應時間的影響。這一工作為建立高效、經濟合成RA甙的酶催化工藝奠定了基礎。
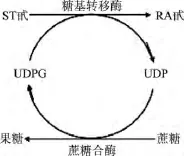
圖1 基于蔗糖合酶的UDPG再生系統(tǒng)Fig.1 Cyclic utilizationof UDPG based on sucrose synthase
1 材料與方法
1.1 試劑與儀器
實驗所用基因和引物 由南京金斯瑞公司合成;CloneEZ PCR Cloning Kit購自南京金斯瑞公司;Taq DNA聚合酶、T4 DNA連接酶、限制性內切酶和2000、15000 DNA Marker 購自Takara公司;質粒抽提試劑盒、DNA片段純化與膠回收試劑盒 購于上海申能博彩有限公司;ST甙和RA甙 購自曲阜海根甜菊制品有限公司。
超聲波細胞粉碎機JY92-IIN 購于寧波新芝生物科技股份有限公司;高速冷凍離心機eppendorf 5810R;ST甙和RA甙濃度測定采用安捷倫1290液相色譜系統(tǒng)和無錫加萊克色譜科技有限公司的NH2柱(4.6 ×250 mm,5 μm 120 ?)。
1.2 菌株和質粒
大腸桿菌菌株E.coli Rosetta(DE3)和載體pET-22b(+)為本實驗室保藏;載體pMDTM-18T 購自TaKaRa公司;質粒pET28a-UGT為本實驗室之前構建的在pET-28a(+)質粒的Nde I和Xho I位點間插入UGT76G1糖基轉移基因的重組質粒。
1.3 培養(yǎng)基
大腸桿菌LB培養(yǎng)基:蛋白胨10 g/L,酵母提取物5 g/L,氯化鈉10 g/L;自誘導培養(yǎng)基(修改的TB培養(yǎng)基[11]):酵母提取物 24 g/L,胰蛋白胨 12 g/L,甘油5 g/L,磷酸氫二鉀12.5 g/L,磷酸二氫鉀2.3 g/L,葡萄糖2 g/L,乳糖3 g/L。
1.4 實驗方法
1.4.1 蔗糖合酶AtSUS1的基因合成 檢索National Center for Biotechnology Information(NCBI)數據庫,根據已公布的擬南芥蔗糖合酶的基因序列(NM_122090),采用 DNAWorks[12]設計 72 條寡核苷酸引物,采用兩步法合成基因[13],如圖2所示。
第一步:將72條引物分為四組,每組18條引物等量混合作為模板,兩端的兩條寡核苷酸作為引物,先合成四個600 bp左右的小片段。PCR采用Primer STAR HS DNA Polymerase(TAKARA)在 50 μL 體系中進行。反應條件為:98℃變性10 s,68℃延伸1 min,30 個循環(huán)。
第二步:重疊PCR。將第一部中合成的四個小片段等量混合作為模板,采用第1條和第72條寡核苷酸引物進行重疊PCR合成大小為2.4 kb的大片段AtSUS1基因。PCR在50 μL體系中進行,反應條件為:98℃變性10 s,68℃延伸2.5 min,30個循環(huán)。
合成的基因用TAKARA公司的DNA產物膠回收試劑盒純化。對純化回收后的基因片段用Nde I和Xho I酶切,對質粒pET-22b(+)采用相同的酶進行雙酶切處理,將以上處理完的表達載體和AtSUS1基因采用T4 DNA Ligase進行16℃過夜連接,次日轉化到E.coli DH5α中涂布于氨芐平板(含氨芐青霉素100 μg/mL)并挑取單克隆,重組質粒為pET22b-SUS,對抽提質粒采用Nde I和Xho I雙酶切鑒定,重組質粒測序工作由南京金斯瑞完成。
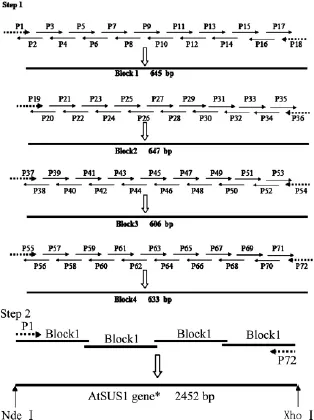
圖2 兩步PCR法合成基因AtSUS1Fig.2 Gene synthesis of AtSUS1 by two-step PCR
1.4.2 雙基因表達質粒 pEUGT-SUS的重組構建 采用CloneEZ?試劑盒將AtSUS1基因亞克隆到質粒pEUGT,得到新的重組質粒pEUGT-SUS,將其轉化到E.coli Rosetta(DE3)中,得到含雙酶基因的重組菌E.coli Rosetta(pEUGT-SUS)。構建過程示意圖如圖3。
以上述pET22b-SUS質粒為模板,用引物P3和P4進行PCR擴增,得到基因AtSUS1。
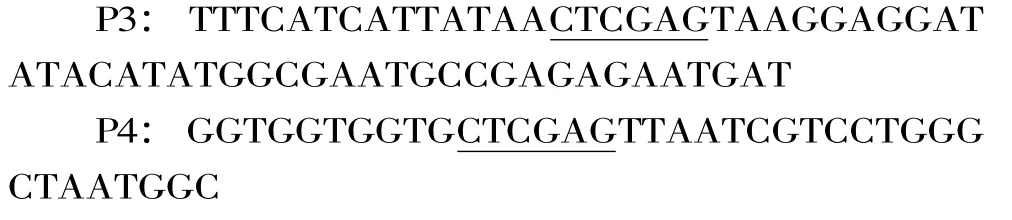
劃線部分為Xho I酶切位點,用相同的酶對質粒pET28a-UGT進行酶切得到線性化載體,用CloneEZ?試劑盒將PCR產物與線性載體進行重組。重組體系為 20 μL:線性化載體 6 μL,PCR 產物 6 μL,10 ×CloneEZ buffer 2 μL,CloneEZ Enzyme 2 μL,去離子水4 μL。得到的表達質粒 pEUGT-SUS導入 E.coli Rosseta(DE3)。
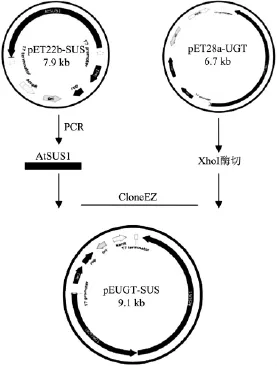
圖3 表達質粒pEUGT-SUS構建過程示意圖Fig.3 Construction of recombinant plasmid pEUGT-SUS
1.4.3 pEUGT-SUS重組菌的誘導表達 挑取重組菌E.coli Rosetta(pEUGT-SUS)單菌落和含pET-28a(+)的對照菌單菌落至5 mL含50 μg/mL卡那霉素和34 μg/mL氯霉素的LB培養(yǎng)基中,37℃,250 r/min培養(yǎng)12 h。然后按2%接種量分別接種到帶相應抗性的自誘導培養(yǎng)基[14]中,37℃培養(yǎng)2 h,轉30℃培養(yǎng)10 h誘導表達。4℃ 8000 r/min離心10 min收集菌體,用0.1 mol/L磷酸鉀緩沖液(pH7.2)懸浮洗滌兩次。用同樣的磷酸鉀緩沖液充分重懸裂解菌體,然后進行超聲破碎(功率300 W,超聲3 s,間歇5 s,共進行10 min)。破碎結束后,4℃ 8000 r/min離心20 min,所得上清液即粗酶液。
1.4.4 糖基轉移酶活性測定 在5 mL的反應體系中 (ST 甙 1mmol/L、UDPG2mmol/L、MgCl21.5 mmol/L、pH7.2),加入1.4.3中的粗酶液進行反應,35℃反應1 h。高溫95℃加熱5 min終止反應,12000 r/min,離心1 min收集上清,然后用高效液相色譜(HPLC)分析RA甙濃度。酶活力單位定義為:在上述反應條件下,每分鐘催化形成1 μmol RA甙所需要的酶量為1個活力單位(U)。
1.4.5 酶催化反應體系 取1.4.3小節(jié)中制備的粗酶液,轉移至小三角瓶中,10 mL反應體系中包括以下物質:ST甙10 g/L、蔗糖10 g/L、粗酶液 82.3 mU/mL、UDP 0.001 mmol/L、MgCl21.5 mmol/L、CaCl210 mmol/L、磷酸鉀緩沖液 0.1 mol/L,pH7.2,35 ℃,反應12 h取樣。高溫95℃加熱5 min終止反應,12000 r/min,離心1 min收集上清,將上清液稀釋10倍,然后用高效液相色譜(HPLC)進行分析。
主要考察了UDP起始濃度、pH、溫度以及反應時間對酶催化反應的影響。在其他條件不變的情況下,分別改變這四個因素。UDP的起始濃度分別為0、0.001、0.01、0.1、0.5、1.0 mmol/L;反應的 pH 分別為6.4、6.8、7.2、7.6、8.0;反應溫度分別為 25、30、35、37、40 和 45 ℃;反應時間分別為 3、6、9、12、16、20、24、30、36 和48 h。
1.4.6 RA甙測定 色譜柱:Lichrospher NH2柱(250 mm ×4.6 mm,5 μm);流動相為乙睛∶水(80∶20,V∶V),用冰醋酸調節(jié)pH至4.56;流速:1 mL/min;柱溫:40℃;檢測波長:210 nm[15]。
2 結果與分析
2.1 AtSUS1基因的合成
采用兩步PCR法合成,第一步先合成四個600 bp左右的小片段,以此為模板進行第二步重疊PCR,合成2.4 kb左右的大片段。PCR產物用0.8%瓊脂糖凝膠電泳鑒定,電泳結果如圖4所示。可知,PCR產物有明顯的特異條帶(泳道2~4為平行制備的三份樣品),對應片段大小與預期結果相符合。
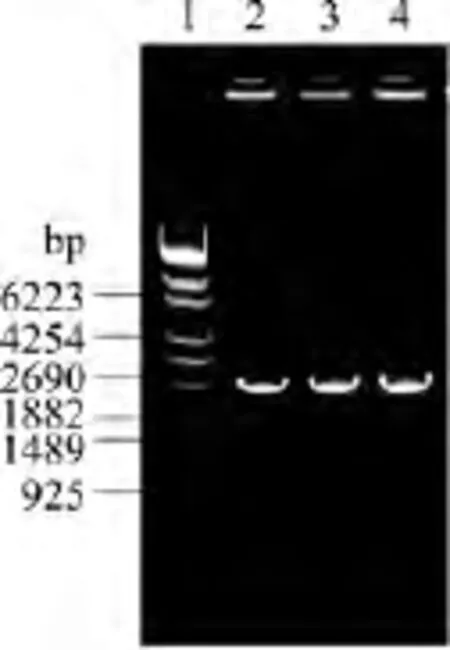
圖4 AtSUS1基因的PCR電泳圖Fig.4 PCR of AtSUS1
回收上述基因片段,采用Nde I和Xho I酶切之后與同樣處理的pET-22b(+)質粒進行連接,連接產物轉化E.coli DH5α中經氨芐平板篩選后,對獲得的陽性克隆提取質粒用Nde I和Xho I進行酶切驗證,結果如圖5所示。在2.4 kb和5.5 kb處出現兩條清晰條帶,分別為目的基因與載體片段對應的大小,說明目的基因已成功連接到質粒pET-22b(+)。通過測序進一步確認目的基因已正確合成,該重組質粒命名為pET22b-SUS。
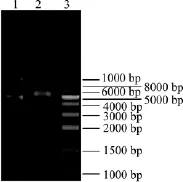
圖5 pET22b-SUS酶切鑒定Fig.5 Enzyme digestion of pET22b-SUS
基因合成技術通過優(yōu)化密碼子和基因修飾為增強異源蛋白表達提供了最有力的工具。本研究中采用了二步PCR法合成了長約2.4 kb的基因片段,在第一步PCR中無需對PCR產物進行純化,其產物直接作為第二步PCR的模板;采用Primer STAR HS DNA Polymerase(TAKARA)在每輪PCR中只需設計兩步不同溫度循環(huán);在引物設計時將酶切位點帶入基因兩端的引物中,獲得的目的片段可經酶切后一步克隆到表達載體上。因此,該方法具有便捷、快速的優(yōu)點。
2.2 表達質粒pEUGT-SUS 的鑒定
用CloneEZ?試劑盒將AtSUS1基因亞克隆到pET28a-UGT質粒。對重組質粒進行Nde I和Xho I雙酶切鑒定,酶切產物經凝膠電泳在2.4、1.4 kb處出現兩條清晰條帶,分別對應AtSUS1和UGT76G1編碼基因的大小,如圖6所示。結果表明目的基因已成功構建到 pET28a-UGT中,所得質粒命名為pEUGT-SUS。
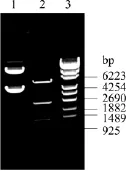
圖6 pEUGT-SUS酶切鑒定Fig.6 Enzyme digestion of pEUGT-SUS
2.3 雙酶催化反應
為了測試蔗糖合酶AtSUS1和UDP糖基轉移酶共表達效果,將重組菌E.coli Rosetta(pEUGT-SUS)表達后制備的粗酶液用于催化反應。分別考察了UDP起始濃度、pH、溫度和反應時間對酶法合成RA甙的影響。
UGT76G1催化的糖基轉移反應,需要UDPG作為活化的糖基供體,而UDPG極其昂貴。蔗糖合酶AtSUS1能將蔗糖與UDP催化生成果糖和UDPG。通過共表達這兩個酶將兩個反應偶聯,該反應體系只需要添加少量相對廉價的UDP就可起始反應,通過蔗糖合酶實現UDPG的再生。分別考察了不同的UDP起始濃度對酶催化合成RA甙的影響,結果如圖7所示。UDP濃度為0~0.01 mmol/L時,隨著 UDP濃度上升,RA甙產量增加;而在此后UDP濃度為0.01~0.5 mmol時,RA甙產量下降,之后幾乎保持不變。在0.01 mmol/L的UDP濃度條件下,10 g/L ST甙轉化生成6.73 g/L RA甙,對應RA甙收率為56.2%。
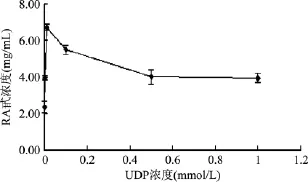
圖7 不同UDP濃度對RA甙產量的影響Fig.7 The influence of different UDP concentration on the yield of RA
分別考察了不同pH緩沖液對酶催化反應的影響,結果如圖8所示,在pH低于7.2時,RA甙生成量較低,這可能是由于在酸性緩沖液中糖基轉移酶的活性較低,且蔗糖合酶反應方向傾向UDPG分解所致[16];當 pH 大于7.2時,RA 甙生成量減少,這可能是由于堿性溶液中Mg2+易沉淀,從而影響酶活性。因此,本反應中緩沖液的最佳pH為7.2。
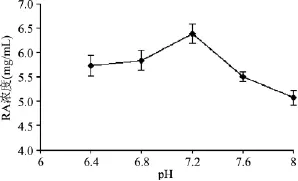
圖8 不同pH對RA甙產量的影響Fig.8 The influence of different pH on the yield of RA
分別考察了不同的反應溫度對酶催化反應的影響,結果如圖9所示,當催化反應溫度小于40℃時,RA甙生成量隨著溫度的升高而增加,說明在25~40℃溫度范圍內,提高溫度對RA甙量有一定促進作用。這可能是溫度的提高,有利于提高糖基轉移酶UGT76G1和蔗糖合酶AtSUS1的活性。在40~45℃,RA甙量逐步減少,說明較高溫度會影響兩個酶的活性。在40℃時,RA甙的濃度最高,為2.36 mg/mL,因此,本反應中最佳溫度為40℃。這一溫度也是重組糖基轉移酶UGT76G1的最適反應溫度[9]。
分別考察了不同的反應時間對酶催化反應的影響,結果如圖10所示,當反應時間小于16 h時,RA甙生成量隨反應時間延長而增加,說明在0~16 h范圍內,隨著反應進行,RA甙的量一直在積累。16 h以后,RA甙濃度開始下降,可能是由于產物RA甙存在一定的降解。在16 h時,RA甙的量達到最高,為6.39 mg/mL,因此,本反應中最佳催化反應時間以16 h。
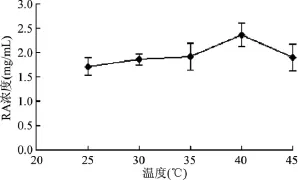
圖9 不同溫度對RA甙產量的影響Fig.9 The influence of different temperature on the yield of RA
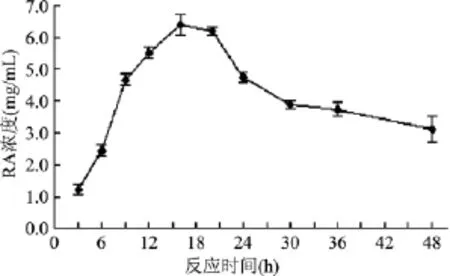
圖10 不同反應時間對RA甙產量的影響Fig.10 The influence of different time on the yield of RA
3 結論與討論
本研究中糖基轉移酶和蔗糖合酶基因均屬植物來源,盡管選用了攜帶稀有密碼子表達質粒的E.coli Rosetta(DE3)作為宿主菌,但重組酶大部分依然以包涵體形式存在,因此,后續(xù)考慮采用融合標簽來增加重組酶的可溶表達。本研究的實驗條件下UDP濃度、反應pH、及溫度等對催化產物合成的影響,是對反應體系中的兩種酶的影響的綜合體現,對糖基轉移酶和蔗糖合酶的反應動力學性質分別表征有利于加深對雙酶系統(tǒng)的了解,進一步提高其轉化效率。
本研究構建了共表達甜葉菊UGT糖基轉移酶和擬南芥AtSUS1蔗糖合酶的重組大腸桿菌E.coli Rosetta(pEUGT-SUS),將重組菌粗酶液用于催化合成RA甙的研究中,初步建立了循環(huán)利用UDPG,催化ST甙合成RA甙的“一鍋雙酶”反應體系,只需要添加相對廉價且遠低于理論值用量的UDP為起始材料。這一策略有利于降低生產成本,推動酶法改質甜菊糖甙的實際應用。
[1]Geigenberger P,Stitt M.Sucrose synthase catalyses a readily reversible reaction in vivo in developing potato tubers and other plant tissues[J].Planta,1993,189:329-339.
[2]Heim U,Weber H,Baumlein H,et al.A sucrose synthase gene of Vicia faba L:expression pattern in developing seeds in relation to starch synthesis and metabolic regulation[J].Planta,1993,191:394-401.
[3]柴靜,俞嘉寧,屈生憲.蔗糖合酶基因AtSUS3干涉后對擬南芥角果發(fā)育的影響[J].西北植物學報,2013,33(4):678-683.
[4]Elling L.Effect of metal ions on sucrose synthase from rice grains-a study on enzyme topograghy[J].Glycobiology,1995,5:201-206.
[5]陳圣,李艷,劉歡,等.生物法合成尿苷二磷酸葡萄糖的研究進展[J].中國生物工程雜志,2012,32(9):125-130.
[6]Masada S,Kawase Y,Nagatoshi M,et al.An efficient chemoenzymatic production of small molecule glucosides with in situ UDP- glucose recycling[J].FEBS Lett,2007,581(13):2562-2566.
[7]Terasaka K,Mizutani Y,Nagatsu A,et al.In situ UDP-glucose regeneration unravels diverse functions of plant secondary product glycosyl transferases[J].FEBS Lett,2012,586(24):4344-4350.
[8]Liu Z,Lu Y,Zhang J,et al.P1 Trisaccharide(Galα1,4Galβ1,4GlcNAc)synthesis by enzyme glycosylation reactions using recombinant Escherichia coli[J].Appl Environ Microbiol.2003,69(4):2110-2115.
[9]劉歡,李艷,嚴明,等.甜葉菊糖基轉移酶UGT76G1的克隆表達及其性質研究[J].食品工業(yè)科技,2012,33(20):187-190.
[10]劉歡,李艷,嚴明,等.重組釀酒酵母全細胞催化合成萊鮑迪甙 A[J].食品與發(fā)酵工業(yè),2012,38(7):6-10.
[11]薩姆布魯克J,拉塞爾DW.分子克隆實驗指南[M].黃培堂,譯.3版.北京:科學出版社,2002:1597.
[12]Hoover DM,Lubkowski J.DNAWorks:an automated method for designing oligonucleotides for PCR-based gene synthesis[J].Nucleic Acids Res,2002,30(10):e43.
[13]Xiong AS,Yao QH,Peng RH.A simple,rapid,high-fidelity and cost-effective PCR-based two-step DNA synthesis method for long gene sequences[J].Nucleic Acids Res,2004,32(12):e98.
[14]Studier FW.Protein production by auto-induction in high density shaking cultures[J].Protein Expr Purif,2005,41(1):207-234.
[15]Kolb N,Herrera JL,Ferreyra DJ,et al.Analysis of sweet diterpene glycosides from Stevia rebaudiana:improved HPLC method.J Agric Food Chem,2001,49(10):4538-4541
[16]Sauerzapfe B,Engels L,Elling L.Broadening the biocatalytic properties of recombinant sucrose synthase 1 from potato(Solanum tuberosum L.)by expression in Escherichia coli and Saccharomyces cerevisiae[J].Enzyme Microb Tech,2008,43:289-296.
