HPLC法測定磺胺甲惡唑鈉有關物質
2015-10-24 02:29:18杜碧瑩
中國醫藥指南 2015年13期
楊 穎 杜碧瑩
(廣州市藥品檢驗所,廣東 廣州 510160)
HPLC法測定磺胺甲惡唑鈉有關物質
楊 穎 杜碧瑩
(廣州市藥品檢驗所,廣東 廣州 510160)
目的 建立高效液相色譜法,測定磺胺甲惡唑鈉有關物質。方法 色譜柱為Zorbax C8(250 mm×4.6 mm,5 μm),流動相為甲醇-磷酸二氫鉀溶液(取磷酸二氫鉀13.6 g,加水至1000 mL,用2%氫氧化鉀溶液調pH值至5.3)(35∶65);檢測波長為210 nm。結果 磺胺甲惡唑鈉與各雜質在該色譜條件下能有效分離,有關物質檢測限0.97 ng。其中雜質F在0.4~5 μg/mL內線性關系良好,r=0.9999 (n =5),定量限2.0 ng。結論 本方法準確,靈敏度高,可用于測定磺胺甲惡唑鈉有關物質。
高效液相色譜法;磺胺甲惡唑鈉;有關物質
磺胺甲惡唑鈉為廣譜抗菌藥,用于治療畜禽、水產類,尿路感染、呼吸道感染、皮膚化膿性感染。原企業標準中磺胺甲惡唑鈉的有關物質檢查采用TLC法,但TLC法存在著諸如薄層板背景干擾大,靈明度不高等缺點,不能有效檢出全部有關物質,不能真實反映該企業樣品的質量狀況。曾有文獻[1]報道用HPLC法測定復方磺胺嘧啶中磺胺甲惡唑鈉的含量。但目前暫未報道磺胺甲惡唑鈉有關物質控制方法。本文建立了
HPLC 法測定磺胺甲惡唑鈉的有關物質,方法簡便、準確、靈敏度高。
1 儀器與試藥
1.1 儀器:日本Shimadzu 高效液相色譜儀LC-10Atvp(二極管陣列檢測器)。
1.2 試藥。雜質A:N-(5-甲基-3-異惡唑基)-4-乙酰氨基苯磺酰胺;雜質F:N-(3-甲基-5-異惡唑基)-4-氨基苯磺酰胺。均為歐洲藥典委員會頒發的對照品。樣品為國外某進口藥品生產廠產品,批號為3010、3009、3008。甲醇為色譜純。水為純化水,其他試劑均為分析純。
2 方法與結果
2.1 色譜條件及系統適用性:色譜柱為ZORBAX C8(250 mm×4.6 mm, 5 μm),流動相為甲醇-磷酸二氫鉀溶液(取磷酸二氫鉀13.6 g,加水至1000 mL,用2%氫氧化鉀溶液調pH值至5.3)(35∶65);流速0.7 mL/min,柱溫為30 ℃,檢測波長為210 nm。取本品和雜質A對照品各1 mg,置10 mL量瓶中,用流動相溶解并稀釋至刻度,搖勻,量取20 μL,注入液相色譜儀,磺胺甲惡唑鈉峰的保留時間約10 min,磺胺甲惡唑鈉峰與雜質A峰的分離度應>3.5。色譜圖見圖1。
2.2 測定方法:取本品,用流動相溶解(45 ℃超聲振搖10 min)并稀釋制成每1 mL中含1 mg的溶液,作為供試品溶液;精密量取適量,用流動相稀釋制成每1 mL中含1 μg的溶液,作為對照溶液(1);另取雜質F對照品適量,精密稱定,用流動相溶解并稀釋制成每1 mL中含1 μg的溶液,作為對照溶液(2)。取供試品溶液、對照溶液(1)、(2)各20 μL,注入液相色譜儀,記錄色譜圖。色譜圖見圖2。
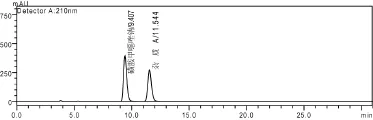
圖1 系統適用性HPLC圖
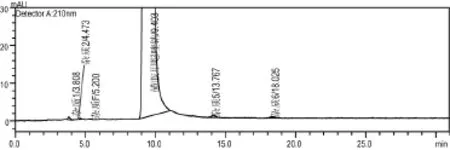
圖2 供試液HPLC圖
2.3 有關物質測定結果,見表1。
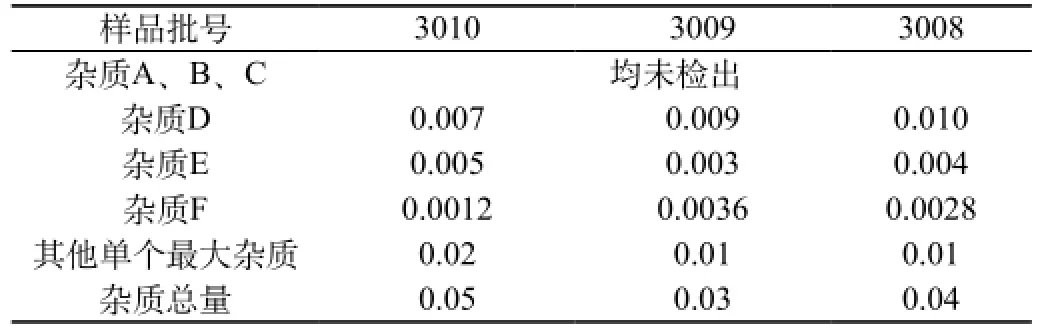
表1 樣品有關物質測定結果
3 方法學驗證
3.1 線性試驗:取本品(批號:3008)、雜質F對照品適量,用流動相分別制成每1 mL中含0.4~5 μg濃度的溶液,依法測定,以濃度為橫坐標,峰面積為縱坐標,計算回歸方程如下:磺胺甲惡唑鈉:Y=86549.5X-1318.11 r=0.9999 (n=5);雜質F:Y=74068.7X+ 6396.62 r=0.9999 (n=5)。
3.2 方法的耐用性試驗:取本品細粉適量,分別通過1 mol/L 鹽酸溶液、1 mol/L 氫氧化鈉溶液、30%過氧化氫溶液放置48 h、254 nm光照4 h和加熱的方式進行破壞,試驗表明,本品遇光易變質,尤其氧化易分解產生降解產物,而設定的色譜條件對雜質有良好的分離。
3.3 進樣的精密度試驗:取對照溶液連續進樣6次,雜質F峰面積變化的RSD(%)為1.4,表明精密度良好。
3.4 檢測限:按信噪比(S/N)為3?1計算,有關物質檢測限0.97 ng,雜質F定量限為2.0 ng。
3.5 供試品溶液的穩定性試驗:在供試品溶液(批號:3008)制備后第0、1、2、4、20、66 h分別測定,供試品溶液在4 h內穩定,隨著放置時間延長至66 h,雜質D(對氨基苯磺酸)與雜質F量略有增加。
4 討 論
4.1 原注冊標準方法為薄層色譜法,影響雜質斑點判定的主要原因是展開劑中的硝基甲烷,硝基甲烷在展開過程中漸變黃色,展開后,黃色附著于薄層板上,直接影響雜質斑點的判定。
從分離效果、靈敏度、專屬性比較,本文所建立的方法靈敏,檢測結果準確且重復性好,綜合破壞性試驗結果,本法能可更好地分離和檢測磺胺甲惡唑鈉的有關物質及貯藏過程的降解產物,故可作為檢查有關物質的色譜條件。
4.2 隨著供試液放置時間延長,雜質D與雜質F量略有增加,因此本品的有關物質檢測應在24 h內進行。
4.3 限度的確定:參照英國藥典[2]“磺胺甲惡唑”有關物質檢查項下的檢測方法及限度要求,結合“磺胺甲惡唑鈉”生產工藝及企業標準綜合評估,我們確定磺胺甲惡唑鈉的雜質包括已知雜質(雜質A、B、C、D、E、F)和未知雜質,雜質F用對照品比較法計算,不得過0.1%;其他雜質用主成分自身對照法計算,不得過0.1%;雜質總量不得過0.3%。結果表明,本方法更準確的反映產品的雜質實際情況,能更好的控制產品的質量。
[1] 武晉孝, 李淑琴, 柴桂珍,等.高效液相色譜法測定復方磺胺嘧啶各組分含量[J].中國獸藥雜志,2002,4(4):30-31.
[2] British Pharmacopoeia Commission.British Pharmacopoeia[S]. The Stationery Office,2013:2127-2129.
Determination of Related Substances of Sodium Sulfamethoxazole by HPLC
YANG Ying, DU Bi-ying
(Guangzhou Institute for Drug Control, Guangzhou 510160, China)
[Abstrat]Objective To establish a method of HPLC to determinate related substances of Sodium Sulfamethoxazole. Methods The analytical column was Zorbax C8(250 mm×4.6 mm,5 μm); The mobile phase was methanol-potassium dihydrogen phosphate solution(consisting 1.36% potassium dihydrogen phosphate, adjust the pH value to 5.3 by 2% potassium hydroxide); the detective wavelength was 210 nm. Results Good resolution of Sodium Sulfamethoxazole and releated substances was obtained, and the detection limit was 0.97 ng. There was a good linear relationship for impurity F in the concentration range of 0.4~5 μg/mL , r=0.9999 (n=5)and the limit of quantification was 2.0 ng. Conclusions The method is accurate, sensitive and suitable for determinaton of related substances of Sodium Sulfamethoxazole.
HPLC; Sodium Sulfamethoxazole; Related substances
R927
B
1671-8194(2015)13-0014-02
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
