超高效液相色譜-串聯質譜法同時測定配方奶粉中泛酸、生物素和氰鈷胺素
2015-11-05 08:32:55李菁菁崔亞娟李全霞張衛民趙茜茜
食品工業科技 2015年12期
李菁菁,葉 潤,崔亞娟,*,李全霞,李 東,路 勇,張衛民,黃 華,趙茜茜
(1.北京市營養源研究所,北京100069;2.北京市食品安全監控和風險評估中心,北京100041)
超高效液相色譜-串聯質譜法同時測定配方奶粉中泛酸、生物素和氰鈷胺素
李菁菁1,葉潤1,崔亞娟1,*,李全霞1,李東1,路勇2,張衛民2,黃華2,趙茜茜2
(1.北京市營養源研究所,北京100069;2.北京市食品安全監控和風險評估中心,北京100041)
建立了超高效液相色譜-串聯質譜聯用技術同時測定配方奶粉中泛酸、生物素和氰鈷胺素的方法。樣品經水溶超聲提取、三氯甲烷除蛋白后,進行超高效液相色譜-串聯質譜分析,采用HSS T3液相色譜柱分離,以10mmol/L乙酸銨溶液(含0.1%甲酸)和乙腈作為流動相進行梯度洗脫,采用電噴霧-正離子電離源多反應監測模式進行定性和定量分析。結果表明:泛酸(VB5)的方法定量限為5μg/100g,生物素(VB7)為2μg/100g,氰鈷胺素(VB12)為0.2μg/100g;回收率為79.4%~87.8%,相對標準偏差為3.3%~5.3%。泛酸的線性范圍0.5~1000ng/mL,生物素為0.2~10ng/mL,氰鈷胺素為0.02~10ng/mL。該方法分析操作簡單、速度快、靈敏度高、重復性好,適用于配方奶粉中泛酸、生物素和氰鈷胺素的同時測定。
泛酸,生物素,氰鈷胺素,超高效液相色譜-串聯質譜法,奶粉
泛酸(Pantothenic acid,VB5)、生物素(Biotin,VB7)、氰鈷胺素(Cyanocobalamin,VB12)是配方奶粉中必須添加的維生素,對嬰幼兒快速發育的各種組織和器官具有十分重要的作用[1]。其含量均較低,尤其是生物素和氰鈷胺素含量通常以微克來表示。
目前,國家標準對配方奶粉中泛酸的檢測方法是微生物法和高效液相色譜法[2]。其中,微生物法[3-5]是測定泛酸的經典方法,但是該方法復雜費時,并對環境條件要求高,影響因素多,且重復性稍差。國家標準對生物素、氰鈷胺素的檢測方法采用微生物法[6-7],具有相類似的問題。高效液相色譜法[8-12]樣品前處理復雜,且雜質峰干擾多,測定基質干擾大、靈敏度低,應用受到限制。國內外利用超高效液相色譜串聯質譜法[13-17]同時測定泛酸、生物素、氰鈷胺素單種或其中2種的相關研究已有報道。
本研究采用基質加標法,可以排除樣品基質的干擾,保證了分析結果的準確性。采用超高效液相色譜-串聯質譜法(UPLC-MS/MS)實現奶粉中泛酸、生物素、氰鈷胺素的同時測定,是一種簡單、快速、準確的分析方法。
1 材料與方法
1.1材料與儀器
奶粉市售配方奶粉;泛酸(VB5,CAS:79-83-4)、生物素(VB7,CAS:58-85-5)、氰鈷胺素(VB12,CAS:68-19-9)純度均大于99.5%,美國IsoSciences公司;甲酸、乙酸銨(均為色譜純) 美國Sigma公司;乙醇、乙腈、三氯甲烷(均為色譜純) 美國Fisher公司;實驗用水超純水。
Acquity@UPLC-Xevo TQ型超高效液相色譜-串聯質譜聯用儀美國Waters公司;Acquity@HSS T3色譜柱美國Waters公司;高速冷凍離心機日本HITACHI公司;漩渦振蕩器美國Scientific Industries公司;分析天平德國Sartorius公司。
1.2實驗方法
1.2.1溶液配制混合標準工作液:精確稱取泛酸鈣、生物素、氰鈷胺素標準品適量(精確至0.01mg)用水溶解,配制成100μg/mL的混合儲備液,冷藏避光保存3個月。臨用時,用水稀釋成標準系列濃度溶液,經0.22μm GHP濾膜過濾,進行UPLC-MS/MS分析。
1.2.2樣品處理稱取樣品1.0g于50mL離心管中,加入10mL水。渦旋振蕩1min,超聲10min。加入10mL三氯甲烷,渦旋振蕩1min,10000r/min離心10min。上清液用超純水適當稀釋,經0.22μm GHP濾膜過濾后上機測定。
1.2.3基質加標實驗提取前加標處理:稱取樣品1.0g于50mL離心管中,分別加入3個不同濃度的混合標準工作液,然后加入10mL水溶解。其后處理同1.2.2。
提取后加標處理:取1.2.2中的上清液1mL于10mL容量瓶中,分別加入3個不同濃度的混合標準工作液。
通過比較提取前加標樣品峰面積(A前)和提取后加標樣品峰面積(A后)得到回收率,扣除了基質效應,回收率計算公式如下:
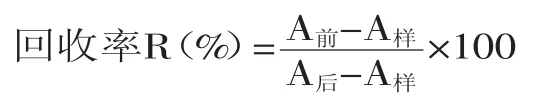
1.2.4色譜條件液相系統:Acquity UPLC系統;色譜柱:Waters Acquity HSS T3柱(1.0mm×100mm,1.8μm);流速:0.2mL/min;柱溫:40℃;樣品溫度:15℃;進樣量:10μL;流動相A:10mmol/L乙酸銨溶液(含0.1%甲酸);流動相B:乙腈;梯度洗脫條件見表1。
1.2.5質譜條件質譜系統:Xevo TQ MS;離子化:ESI+;監測方式:多反應檢測(MRM);毛細管電壓:2.3kV;離子源溫度:150℃;脫溶劑氣溫度:500℃;錐孔氣流量(氮氣):50L/h;脫溶劑氣流量(氮氣):1000L/h;碰撞氣流速(氬氣):0.17mL/min。3種維生素的定量和定性離子對、錐孔電壓、碰撞能量等參數見表2。

表1 流動相梯度洗脫條件Table 1 Elution condition of the thirteen sedatives
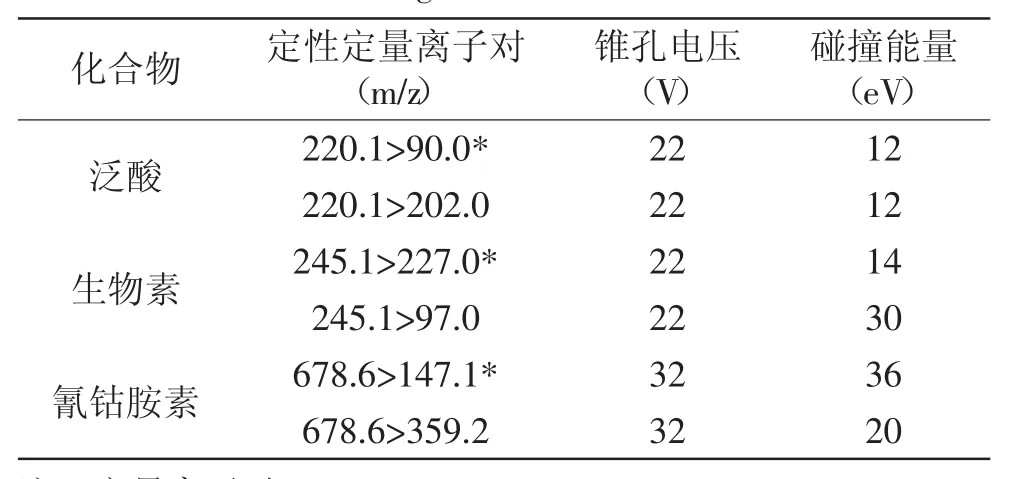
表2 3種維生素的定性和定量離子對、錐孔電壓和碰撞能量Table 2 Confirmation and quantitative ions,cone voltages and collision energies of the three vitamins
2 結果與分析
2.1提取條件選擇
針對泛酸、生物素、氰鈷胺素既能溶于水,又能微溶于乙醇的性質,分別用水、10mmol/L乙酸銨和乙醇作為樣品提取溶劑作為提取劑進行實驗。結果表明:選擇乙醇作為提取劑,可同時提取出奶粉中的其他有機物質,增加了基質的干擾;選擇水和10mmol/L乙酸銨作為提取劑,靈敏度高、峰形穩定,效果最好。考慮到盡量簡化提取操作的原則,選擇水作為提取劑。
另外,針對奶粉樣品中高蛋白的特性,分別考察了高氯酸和三氯甲烷的去除蛋白效果。結果表明,2種方式都可以使蛋白沉淀,獲得澄清的樣品溶液。但高氯酸對3種生物素的破壞性較大,處理后樣品的響應值明顯下降。而三氯甲烷對回收率無明顯影響。因此選擇三氯甲烷作為除蛋白劑。
2.2基質效應考察
基質是樣品中除分析物以外的組分,對分析物的分析有顯著干擾,并影響分析結果的準確性,這些影響和干擾被稱為基質效應(ME)。LC-MS/MS中的基質效應由分析物的共流出組分影響電噴霧接口的離子化效率所致,表現為離子增強或抑制作用[18]。
實驗采用提取后添加法建立數學模型評定基質效應。基質效應(ME)=Set1/Set2,其中Set1:純的標準品溶液信號峰面積值,Set2:樣品基質提取后添加信號峰面積值。2個條件下重復測定5次。結果表明ME值均<60%,奶粉樣品中基質復雜,基質效應不可忽略。
2.3色譜條件選擇
水溶性維生素通常很難在反相色譜柱上用常規的方法分離。本實驗選用T3色譜柱,分別采用10mmol/L乙酸銨溶液(含0.1%甲酸)和純水、乙腈與甲醇作為流動相,以不同比例梯度進行洗脫。結果表明,流動相選用10mmol/L乙酸銨溶液(含0.1%甲酸)和乙腈梯度洗脫,泛酸、生物素、氰鈷胺素達到較好的分離效果,出峰時間分別為2.21、2.67、2.49min。
2.4質譜條件選擇
使用UPLC-MS/MS方法,通過多反應監測(MRM)方式進行采集,選擇目標物質的兩對母離子/子離子,其中響應較強的離子對作為定量離子對,另一對作為定性離子對。3種維生素的MRM色譜圖見圖1。在電噴霧源正離子檢測(ESI+)方式下,3種維生素的準分子離子峰為[M+H]+峰。實驗調節各個參數得到響應較強的[M+H]+母離子峰。然后對母離子進行子離子掃描,調節碰撞能量。優化確定適合本實驗條件的質譜條件,見表2。

圖1 3種維生素的定量離子色譜圖Fig.1 The quantification ion chromatogram of the three vitamins
2.5方法的驗證
2.5.1回收率分別添加3個不同質量濃度(表3)標準溶液進行提取測定,泛酸、生物素、氰鈷胺素的平均回收率分別為79.4%、84.8%、87.8%,RSD分別為4.4%、5.3%、3.3%。表明本法準確性較好,適合配方奶粉中3種維生素的定量分析。
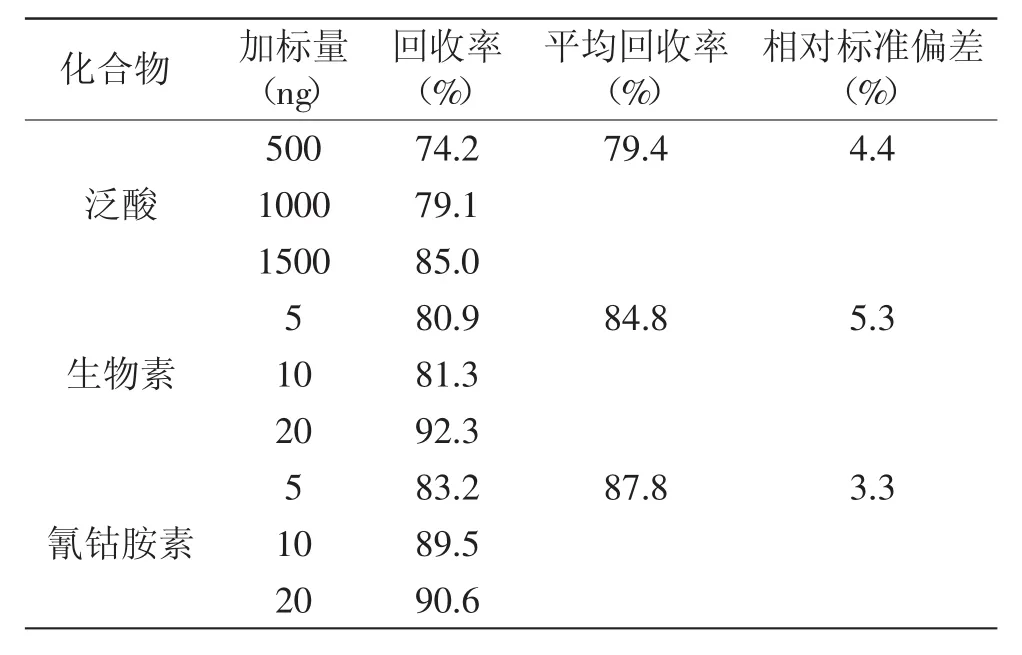
表3 3種維生素添加回收實驗結果(n=6)Table 3 Average recoveries and relative standard deviation of the three vitamins(n=6)
2.5.2線性范圍、檢出限和定量限取1.2.2中的上清液1mL至10mL容量瓶中,添加100μL含3種分析物的已知濃度標準溶液,加水定容,進樣分析。得到相關系數≥0.9985的較好線性關系,如圖2所示,泛酸的線性范圍0.5~1000ng/mL,生物素為0.2~10ng/mL,氰鈷胺素為0.02~10ng/mL。
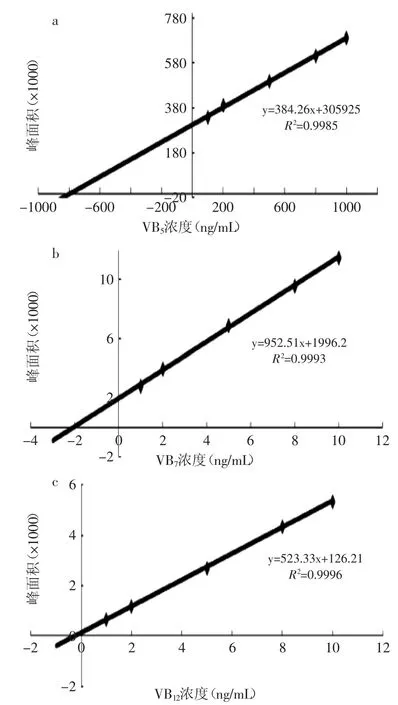
圖2 3種維生素的校正曲線Fig.2 The calibration curve of the three vitamins
根據各定量離子3倍信噪比(S/N)對應樣品中的濃度得到方法檢出限,結果見表4。泛酸、生物素、氰鈷胺素檢出限分別為2、0.6、0.06μg/100g;以10倍信噪比(S/N)對應樣品中的濃度得到方法定量限,泛酸、生物素、氰鈷胺素定量限分別為5、2、0.2μg/100g。實驗表明可以對3種維生素含量較低的樣品進行定量分析。

表4 3種維生素的回歸方程、檢出限和定量限Table 4 Regression equations,limits of determination and limits of quantification of the three vitamins
2.5.3方法精密度取奶粉樣品按照1.2.2中的方法處理,平行測定6次(n=6),考察方法的精密度,測定結果見表5。泛酸、生物素、氰鈷胺素測定的相對標準偏差在3.1%~5.1%,表明該方法精密度較好,適用于奶粉中低含量的生物素、氰鈷胺素的定量分析。

表5 3種維生素的測定結果Table 5 The result of determination of the three vitamins
3 結論
3.1以水溶解并超聲提取配方奶粉中的水溶性維生素,并采用三氯甲烷除去提取液中蛋白,用0.22μm GHP濾膜過濾后進行測定。該操作簡單,分析周期短,不需要復雜的前處理,得到了較好提取效果。
3.2通過基質加標法測定泛酸、生物素、氰鈷胺素等三種維生素,樣品回收率滿足要求,表明該方法可用于同時測定上述3種物質的含量。本方法可替代使用價格昂貴的同位素內標法,降低中小企業的檢驗成本。
3.3建立了超高效液相色譜-串聯四級桿質譜(UPLC-MS/MS)測定方法,采用電噴霧-正離子電離源多反應監測模式進行定性和定量分析,將檢測時間控制在5min之內,大大提高了檢測效率,值得推廣。
[1]張丹參,杜冠華.維生素營養與健康基礎(第三版)[M].北京:科學出版社,2009:3-6.
[2]中華人民共和國衛生部.GB 5413.17-2010食品安全國家標準嬰幼兒食品和乳品中泛酸的測定[S].北京:中國標準出版社,2010.
[3]李全霞,崔亞娟,趙寅菲,等.微生物法測定食品中水溶性維生素的原理及進展[J].食品科學,2013,34(13):338-344.
[4]Angyal G.Methods for microbiological analysis of selected nutrients[J].USA:AOAC International,1996,8(1):41-45.
[5]張旭,馬妮,鄭洪.微生物法測定食品中泛酸的含量[J].中國微生態學雜志,2012,24(7):654-655.
[6]中華人民共和國衛生部.GB 5413.14-2010食品安全國家標準嬰幼兒食品和乳品中維生素B12的測定[S].北京:中國標準出版社,2010.
[7]中華人民共和國衛生部.GB 5413.19-2010食品安全國家標準嬰幼兒食品和乳品中游離生物素的測定[S].北京:中國標準出版社,2010.
[8]周迅雷,張志國,褚慶環,等.食品中VB12檢測方法研究進展[J].食品與發酵工業,2008,34(11):131-134.
[9]杜彥山,張志國,賈云虹,等.高效液相色譜法測定奶粉中泛酸[J].食品研究與開發,2007,128(6):121-124.
[10]Pilar V,Carmenl E,Nuri A B,et al.Reversed-phase liquid chromatography on an amide stationary phase for the determination of the B group vitamins in baby foods[J].Chromatogr A,2003,1007:77-84.
[11]Markopoulou C K,Kagdadis K A,Koudourellis J E.An optimized method for the simultaneous determination of vitamins B1,B6,B12in multivitamin tablets by high performance liquid chromatography[J].Journal of Pharmaceutical and Biomedical Analysis,2002,30:1403-1410.
[12]李少旦,彭衛芳.反相高效液相色譜法同時測定維生素B6、煙酰胺和泛酸鈣[J].理化檢驗-化學分冊,2009,45(7):800-802.
[13]渠巖,崔亞娟,李全霞,等.超高效液相色譜-同位素稀釋質譜法測定奶粉中的泛酸[J].食品科學,2014,35(8):212-215.
[14]林宏琳,華永有,黃宏南.液相色譜-串聯質譜法測定保健食品中維生素B12[J].中國食品衛生雜志,2011,23(5):432-434.
[15]劉進璽,鐘紅艦,董小海.超高效液相色譜-質譜聯用測定維生素預混合飼料中生物素含量[J].分析實驗室,2010,29(7):62-64.
[16]Michael.Simultaneous analysis of folic acid and pantothenic acid in foods enriched with vitamins by stable isotope dilution assays[J].Analytica Chimica Acta,2003,495:133-141.
[17]Michael R,Achim F.Quantification of pantothenic acid and folates by stable isotope dilution assays[J].Journal of food composition and analysis,2002,15:399-409.
[18]向平,沈敏,卓先義.液相色譜-質譜分析中的基質效應[J].分析測試學報,2009,28(6):753-756.
Determination of pantothenic acid,biotin,cyanocobalamine in formula milk powder using ultra performance liquid chromatography-tandem quadrupole mass spectrometry
LI Jing-jing1,YE Run1,CUI Ya-juan1,*,LI Quan-xia1,LI Dong1,LU Yong2,ZHANG Wei-min2,HUANG Hua2,ZHAO Xi-xi2
(1.Beijing Research Institute for Nutritional Resources,Beijing 100069,China;2.Beijing Municipal Center for Food Safety Monitoring and Risk Assessment,Beijing 100041,China)
Ultra performance liquid chromatography-tandem quadrupole mass spectrometry(UPLC-TQMS)method had been developed for the determination for pantothenic acid(VB5),biotin(VB7),cyanocobalamine(VB12)in formula milk powder.The samples were ultrasonic extracted by water,and precipitated protein by chloroform for analyzing by UPLC-MSMS.The analytes were separated by HSS T3 column.The mobile phase consisted of 10mmol/L ammonium acetate with 0.1%formic acid and acetonitrile.The samples were identified and quantified by multiple reaction monitoring(MRM)via positive electrospray ionization(ESI+).The results showed that the limit of quantification of VB5were 5μg/100g,VB7were 2μg/100g,VB12were 0.2μg/100g.The recoveries at three spiked levels and the relative standard deviations were 79.4%~87.8%and 3.3%~5.3%,respectively.This method was simple,time saving,sensitive and accurate in the determination of VB5,VB7,VB12in formula milk powder.
pantothenic acid;biotin;cyanocobalamine;ultra performance liquid chromatography-tandem quadrupole mass spectrometry;milk powder
TS207.3
A
1002-0306(2015)12-0061-04
10.13386/j.issn1002-0306.2015.12.004
2015-02-02
李菁菁(1985-),女,碩士研究生,研究方向:食品科學與工程。
崔亞娟(1979-),女,碩士研究生,研究方向:分析與檢測。
北京市科學技術研究院青年骨干計劃(201315);北京市科技計劃(Z141100002614013)。
