基于氫鍵的陰離子識別主體分子的研究進展
2015-12-01 02:36:33李勇軍劉輝彪李玉良
無機化學學報 2015年9期
李勇軍劉輝彪 李玉良
(中國科學院化學研究所有機固體院重點實驗室,北京100190)
基于氫鍵的陰離子識別主體分子的研究進展
李勇軍*劉輝彪李玉良
(中國科學院化學研究所有機固體院重點實驗室,北京100190)
陰離子在生物學、醫學、催化以及環境等方面逐漸得到廣泛認識與重視,陰離子受體研究在跨膜離子輸運、化學傳感、模擬酶催化有機化學反應等方面亦有光明的應用前景。本文根據酰胺、脲與硫脲、吲哚吡咯、三氮唑、銨鹽、胍鹽、咪唑、羥基等不同的氫鍵單元,總結基于氫鍵的陰離子識別主體分子的研究進展。
陰離子識別;主體分子;氫鍵
0 引言
1968年杜邦公司的Park和Simmonds報道了一系列大二環主體分子1的鹵素陰離子的絡合性質[1]。當該大二環的橋頭氮原子質子化后,鹵離子可以結合到其大二環的空腔內。并通過X射線晶體結構的測定得以確定,這成為陰離子被大環主體結合的第一例。而就在幾乎同期,Pedersen報道了他的劃時代結果,關于二苯并[18]冠6配位陽離子的行為[2],這兩大劃時代的成果標志著現代超分子化學的開始。
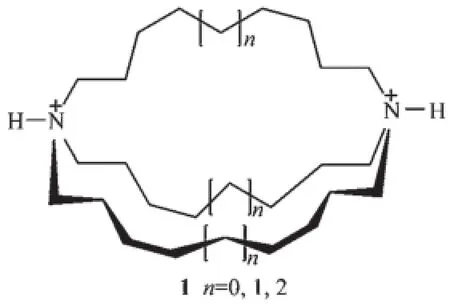
圖1 大二環主體分子1的結構Fig.1 Molecular structure of macrobicyclic amine 1
雖然鍵合陰離子和陽離子的人工合成受體分子幾乎是在同一時間被發現的,但是與陽離子和中性分子主體相比較,非共價鍵陰離子配位化學的發展速度則較為緩慢,這主要是由于:陰離子半徑相對較大,要求受體比陽離子受體的半徑要大。陰離子的形狀幾何構型多樣,有球形(鹵離子)、線形(SCN-、N3-),平面(NO3-、PtCl42-)、四面體(PO43-、SO42-)、八面體(PF6-、Fe(CN)63-),以及生物體中的更加復雜的例子如低聚磷酸根陰離子等。陰離子與陽離子比較,有更高的溶劑化自由能,因而陰離子主體會與周圍介質發生競爭。許多陰離子還受到pH值的影響,只能穩定存在于相對較窄的pH值范圍內,尤其是多銨鹽的受體。陰離子配位通常是飽和的,只能通過弱作用力(氫鍵和范德華相互作用)結合。所以,陰離子客體的選擇性結合要比金屬陽離子的結合存在更多更大的挑戰。
在20世紀的70、80年代,陰離子配位化學發展一直比較緩慢,直到80年代末,陰離子配位才真正受到關注[3-6]。化學家們設計合成了大量基于分子間弱相互作用的新型陰離子主體分子,這些分子無論從設計合成、構造尺度以及陰離子的配位方式上都顯示著化學家的智慧[7]。氫鍵由于其方向性強,種類多樣且易于修飾,基于氫鍵可以設計出特定結構的受體分子來識別不同空間構型及氫鍵接受能力的陰離子[7-8]。本文將根據不同的氫鍵單元,總結基于氫鍵的陰離子識別主體分子的研究進展。
1 基于酰胺的陰離子識別主體分子
1986年,Pascal[9]合成了第一個純酰胺的陰離子識別主體化合物2。在DMSO溶液中,該化合物表現了對氟離子識別的特性。隨后,大量的酰胺類陰離子受體分子涌現出來[10-14]。Kubik教授研究組設計合成了一類環肽的陰離子識別主體化合物3[15-16],能夠與碘離子或硫酸根離子在水溶液中形成1∶1的復合物,其中3e表現出與硫酸根強的作用力,lgKa= 5.97,成為水溶液中良好的中性受體分子。
Chmielewski和Jurczak研究組合成了一類含有吡啶基團的四酰胺的環狀化合物4[17-19],并研究了它們與陰離子配位的性質。發現含有吡啶基團的酰亞胺比間苯二甲酰亞胺與陰離子的作用要強,而且環狀與非環狀相比較,與陰離子作用更強。Lin教授研究組合成了化合物5[20-21],該化合物表現出對氟離子的良好的選擇性。由于與氟離子形成了氫鍵,化合物的顏色發生變化。值得一提的是這種作用不是由于氟離子的去質子化作用引起的,因為同樣的四丁基氫氧化銨就沒有這種效果。Bowman-James和Hossain教授研究組設計合成了一種多功能的主體分子6,可以同時配位陰離子和陽離子客體[22-24]。6的質子化形式H262+形成折疊結構來配位Cr2O72-或ReO4-,中間的質子化的胺氫由于氫鍵作用向上翻,從而使得含氧陰離子能夠與里面的酰胺作用。進一步質子化后,能夠與金屬陽離子發生配位作用,而且作用方式與陰離子相似。Kim教授組設計合成了含芘的杯[4]冠5陰離子識別主體分子7[25-26],該分子通過冠醚基團可以配位K+,這種結合改變了主體分子的構象,從而增大了芘分子之間的π-π作用,最終導致芘熒光增強。而在陰離子H2PO4-加入時,由于PET效應,芘的熒光被淬滅。據此,該分子可以被設計成為一個inhibit(INH)的邏輯門開關。
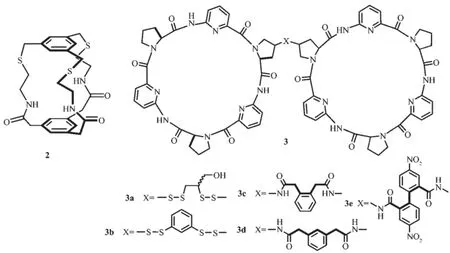
圖2 基于酰胺的陰離子識別主體分子2和3的結構Fig.2 Molecular structure of amide-based anion receptors 2 and 3
Bowman-James教授研究組合成了一系列環蕃類的陰離子識別主體化合物8a~e[27],該類主體分子外形像一個口袋,通過酰胺與陰離子的作用,可以用來配位識別陰離子。
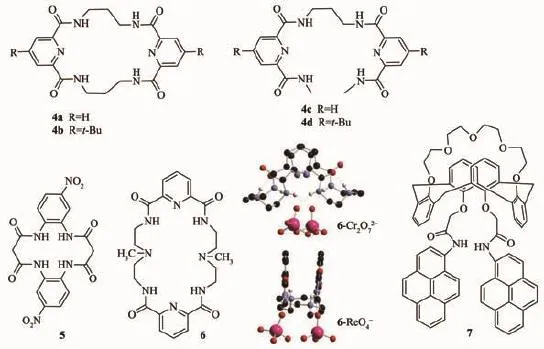
圖3 基于酰胺的陰離子識別主體分子4~7的結構以及6與Cr2O72-和ReO4-的配合物晶體結構Fig.3 Molecular structure of amide-based anion receptors 4~7,and the crystal structures of complexes of 6 with Cr2O72-and ReO4-
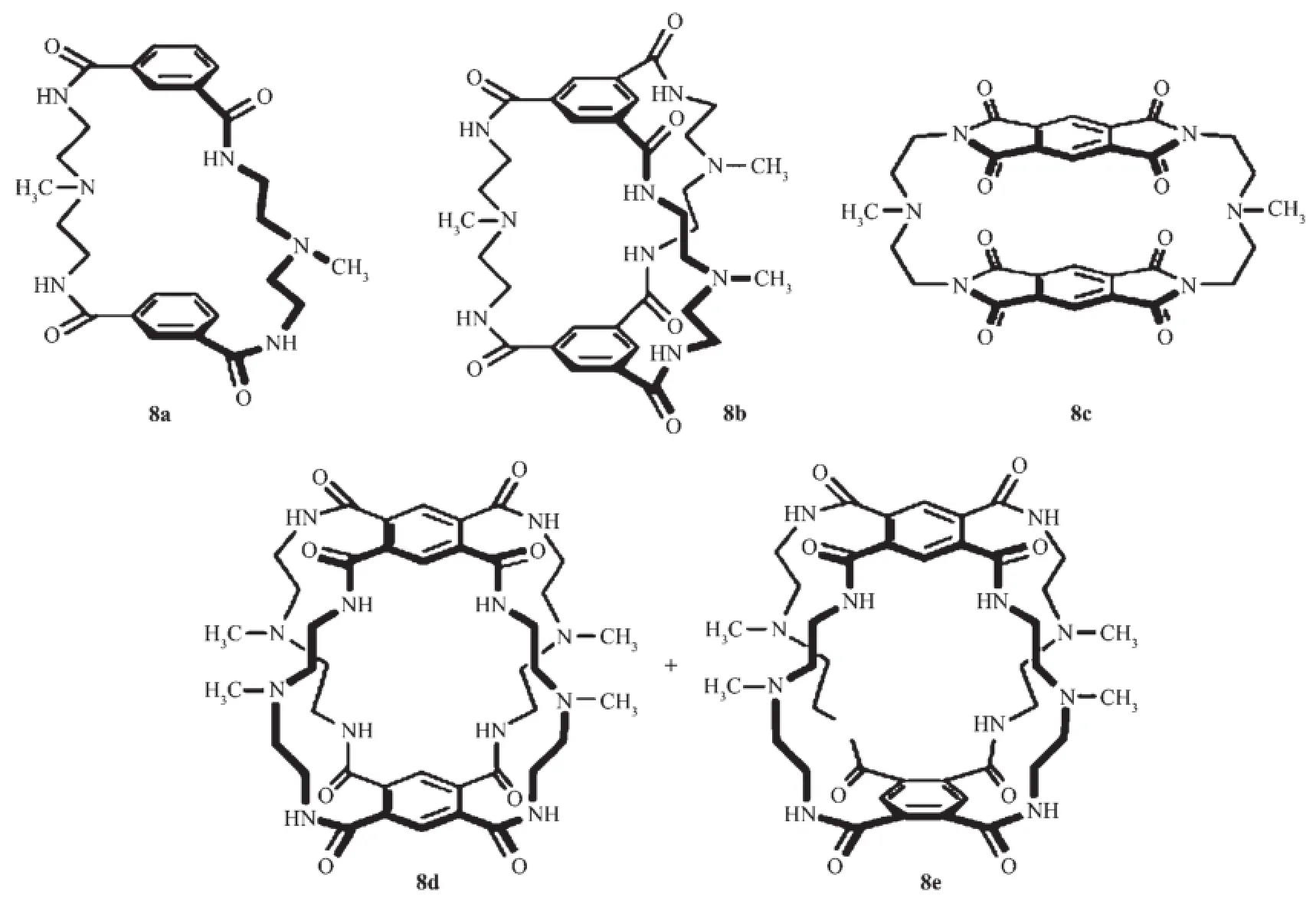
圖4 基于酰胺的陰離子識別主體分子8a~e的結構Fig.4 Molecular structure of amide-based anion receptors 8a~e
酰腙單元具有類似于酰胺的NH氫鍵給體,但它的可逆性使得它可以用于動態組合化學。Beeren和Sanders教授采用這種策略設計了一種優秀的磷酸二氫根受體[28]。獲得了腙連接的基于二茂鐵的大環和線性低聚物動態組合庫,并且發現四丁基磷酸二氫銨的加入導致大環化合物產率降低而線性受體9的產率急劇增加。9與磷酸二氫根采取2∶1的結合模式,在亞胺NH,環戊酰腙CH和NH氫鍵的正協同效應下采取螺旋構象。與酰胺類似作用方式的方胺也被用于陰離子識別,并且Gale等人將其通過類固醇結構組裝為10,實現了高達1014mol-1·L的氯離子親和力[29]。

圖5 基于酰腙或方胺的陰離子識別主體分子9與10的結構Fig.5 Molecular structure of hydrazone-based receptor 9 and squaramide-based anion receptor 10
2 基于脲與硫脲的陰離子識別主體分子
脲和硫脲是兩類非常好的氫鍵給體化合物,對于含氧陰離子來說,它們是非常出色的識別主體。Peng教授研究組合成了一類簡單的具有激發態分子內電荷轉移(ESIPT)性質的含有脲的陰離子受體11[30]。熒光放射光譜包含2個峰,短波長(414 nm)和長波長(554 nm)。長波長的是由于(ESIPT)效應引起的,當加入陰離子后,構象改變,ESIPT效應被抑制,熒光光譜發生藍移。
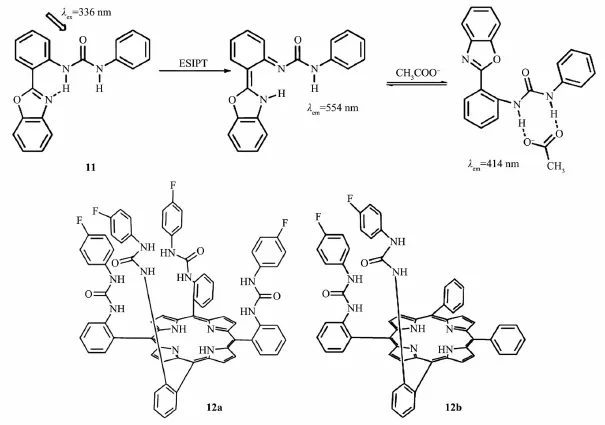
圖6 基于脲的陰離子識別主體分子11和12的結構及11對陰離子的識別機理Fig.6 Molecular structure of urea-based anion receptors 11 and 12,and anion sensing mechanism of 11
Burns教授研究組合成了基于卟啉和脲的陰離子識別主體[31-32],溶劑分子可以影響其對于陰離子的選擇性。當有DMSO溶劑分子存在時,12a表現出對Cl-高度的選擇性,這是因為DMSO能夠把Cl-穩定在脲的附近。而對于體積較大的H2PO4-和COO-來說,溶劑的效應就不那么明顯。12b少2個脲基團因此不能包溶劑分子,所以與Cl-的結合就比較弱,從而證明了溶劑在識別陰離子過程中的作用。
Ghosh教授研究組合成了分子10a[33],Das和Ganguly教授合成了類似的分子10b[34]。其中10a表現出與H2PO4-、AcO-良好的配位能力。lgKa分別為5.52和4.45。10b表現出對SO42-、H2PO4-良好的選擇性,lgKa分別為4.97和4.26。
Gozin教授研究組合成了14a、14b兩種分子[35]。并比較了它們與陰離子的配位性質,14a結合常數的順序為H2PO42->F->AcO-,而14b的順序為AcO-> F->H2PO42-,正是由于空腔大小的原因,14b對體積較小的陰離子表現出良好的配位能力,而14a則表現出對于大體積陰離子的識別優勢。Gale教授研究組合成了15a、15b兩種基于酰胺與脲的陰離子受體分子[36-37],15a表現出與AcO-強的配位作用。而15b在溶液中不穩定,易水解。而且也沒有表現出類似于15a那樣的陰離子的配位性質,原因是15b以彎曲構象存在,容易水解。而15a中吡啶氮原子能與旁邊的酰胺形成氫鍵從而防止分子形成彎曲構象。Bhmer教授研究組用陰離子模板的方法合成了一系列的含有脲的大環化合物16a、126[38-40],1H NMR研究證明隨著苯環個數的增多,大環的柔韌性變好,與陰離子的配位常數變小。
同樣,Custelcean等合成了對稱的聯吡啶取代脲配體17并以鋅(Ⅱ)配位構建M4L6籠[41]。希望M4L6籠的空腔有利于四面體陰離子的配位。利用核磁共振技術監測了配合物在溶液中的形成。往17的2∶1 CD3OD∶D2O溶液加入硝酸鋅,6個配體和4個鋅(Ⅱ)離子在幾分鐘內形成低對稱性配合物或混合配合物。而隨著EO42-陰離子(E=S,Se,Cr,Mo,W,P)的加入,形成了預期的Zn4L6籠。吳彪等報道了強烈結合硫酸根的三足六脲配體18[42]。作者發現,在固態時1個單一的SO42-陰離子可以通過12個氫鍵被6個尿單元所包裹,使得18是第一例能完全由單一有機主體分子滿足SO42-陰離子的飽和配位數的分子。該結構采用四面體籠,3條臂的每個終端沿著底部三角面的邊緣折疊,每個尿單元配位四面體陰離子的一個邊。作者還研究了溶液相的陰離子結合性能,發現其在強競爭溶劑DMSO-d6-H2O 25%中的穩定常數Ka>104mol-1·L。也發現受體18能夠有效地從NaNO3-Na2SO4溶液中提取SO42-陰離子。
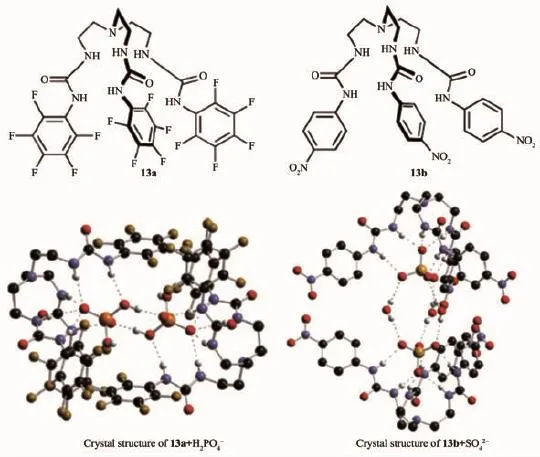
圖7 基于脲的陰離子識別主體分子13a與13b的結構及配位H2PO4-和SO42-的晶體結構Fig.7 Molecular structure of urea-based anion receptors 13a and 13b,and the crystal structures of 13a+H2PO4-and 13b+SO42-
Feringa教授提出了一種基于第一代分子馬達的雙脲受體19。該受體對磷酸二氫根具有很強配位能力,并且可以利用光化學和熱驅動分子在3個異構體之間切換,表現不同的陰離子結合力。這提供了前所未有的配位底物三級控制能力。此外,這也是迄今為止最有效的含氧陰離子的光控受體分子。這在光控含氧陰離子的輸運、光控藥物釋放和跨膜離子輸運的調控等方面具有潛在應用[43]。
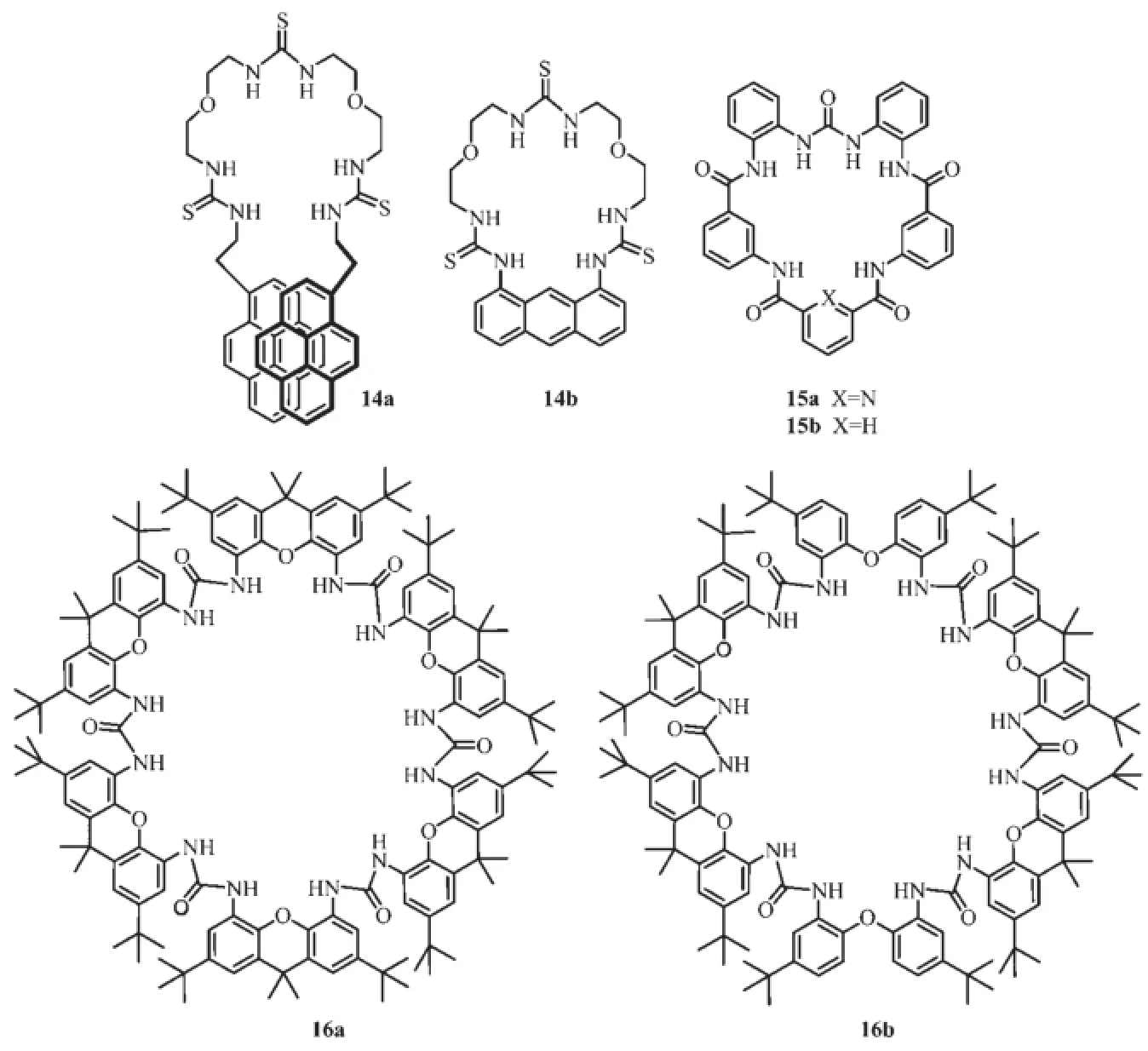
圖8 基于脲和硫脲的陰離子識別主體分子14、15與16的結構Fig.8 Molecular structure of urea/thiourea-based anion receptors 14,15 and 16

圖9 基于脲的籠裝陰離子識別結構Zn4(17)6及18-SO42-的晶體結構Fig.9 Crystal structures of cage-like urea-based anion receptor system Zn4(17)6and 18-SO42-

圖10 光和熱驅動的雙脲受體19Fig.1 0Light-and heat-responsive bis-urea receptor 19 towards H2PO4-
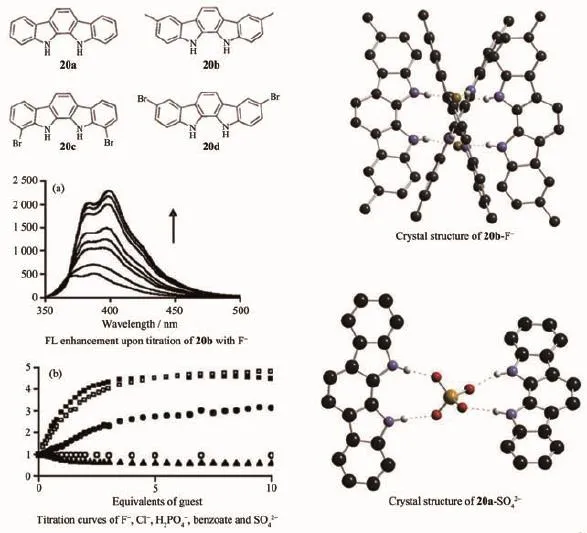
圖11 基于吲哚的氮茚并氮芴的陰離子識別主體分子20a~d的分子結構、熒光光譜滴定、配合物晶體結構Fig.1 1Molecular structure of indolocarbazoles-based anion receptors 20a~d,fluorescent titration and crystal structures of 20b-F-,20a-SO42-
3 基于吲哚及吡咯的陰離子識別主體分子
吡咯和吲哚都含有一個氫鍵給體,能夠配位陰離子,但是吡咯作為陰離子配位主體單元已經被研究的很多[44],而吲哚則是一個新型的陰離子識別主體單元[45]。而且與吡咯相比,吲哚的酸性要強,所以能夠產生與陰離子更強的氫鍵作用。2005年,牛津大學的Beer教授報道了第一例基于吲哚的氮茚并氮芴的化合物,并研究了其與陰離子的配位性質[46]。如圖,化合物20a~d,在陰離子引入時,紫外熒光光譜都發生改變,尤其在F-、H2PO42-、Cl-引入時,熒光強度發生明顯的增大。從化合物20b與F-的單晶結構中可以看出,4個化合物20b分子圍繞在2個F-周圍,形成一種螺旋型的結構。最近他們研究了該類分子與SO42-的作用,發現該類化合物20a能夠與SO42-形成2∶1的結構[47],這些研究都預示著基于吲哚的氮茚并氮芴化合物在未來陰離子模板組裝中有著巨大的潛力。
Jeong教授研究組合成了一系列的基于吲哚單元的氮茚并氮芴環狀的陰離子識別主體分子21a~d。這些剛性的大環根據其空腔的大小可以用來識別不同的陰離子。環21a表現出與F-強的配位作用[48],結合常數為5.6×108mol-1·L。而與Cl-的結合常數為2.1×106mol-1·L。21a與Cl-的單晶結構顯示Cl-位于大環的一側。在21a、21b與N3-配位的過程中,對于較小的環21a,N3-僅端點與環發生氫鍵作用,而在較大的環21b中,N3-整個占據在空腔內部,兩端的N原子均形成氫鍵作用[49]。而且后者比前者更加穩定,結合常數分別為2.3×103、8.1×104mol-1·L。在化合物21d中,由于吡啶的存在使得其與H2PO42-、Cl-配位后表現出不同的構象[50],在H2PO42-存在時,形成吡啶氮原子與H2PO42-中氫原子的氫鍵,而Cl-存在時,吡啶中的C-H原子與Cl-形成氫鍵。化合物21c[51]與AcO-、H2PO42-、Cl-的結合常數分別為1.1×106、2.9×104、5.6×104mol-1·L。在單晶結構中,化合物21c與H2PO42-形成了2∶2的復合物。我們基于吲哚衍生物氮茚并氮芴和三氮唑設計合成環狀陰離子受體分子22、23[52],通過識別基團平面之間存在的π-π作用,可以自組裝成一種折疊結構,形成的空腔又可以通過氫鍵作用來識別陰離子客體。其中環23與陰離子形成了夾心配合物結構。
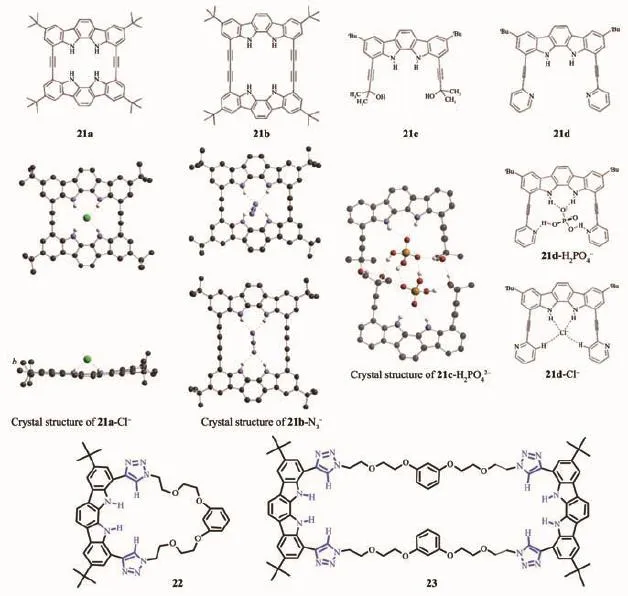
圖12 基于吲哚的氮茚并氮芴的陰離子識別主體分子21,22與23的結構及21與陰離子配位的晶體結構Fig.1 2Molecular structure of indolocarbazoles-based anion receptors 21,22 and 23,and the crystal structures of 21 with anions
Gale教授研究組合成了一類“V”字形的含有吲哚基團的陰離子識別主體分子,24a、24b均表現出對于F-良好的選擇性,單晶結構顯示在F-存在時,24b是以一種扭曲的形式配位,而體積更大的Cl-位于主體分子的一側。正式由于這種結合方式的不同,主體分子才表現出對F-良好的選擇性[53]。同時該研究組也合成了一系列的含有吲哚和脲的陰離子識別主體分子24c、24d[54-55]、24e、24f[56]。其中,24d表現出對于H2PO4-的選擇性。而在晶體結構中,H2PO4-發生了去質子化作用,以PO43-的形式存在。同樣HCO3-與24c的晶體結構中,HCO3-也發生了去質子化作用,以CO32-的形式存在。24e、24f能夠與含氧陰離子形成能夠六氫鍵的配合物,1H NMR滴定研究表明,在AcO-滴定過程中,形成了脲與吲哚的4個氫鍵,而在H2PO4-、HCO3-的存在下形成了全部的6個氫鍵。而且隨著H2PO4-加入量的增多,發生去質子化作用,最終形成HPO42-的配合物。

圖13 基于吲哚的陰離子識別主體分子24a~f的結構及與陰離子配位的晶體結構Fig.1 3Molecular structure of indole-based anion receptors 24a~f,and the crystal structures of 24 with anions
Sessler和Lee教授研究組合成了一類杯[4]吡咯25a~d[57-58],他們與Hey教授合作,比較了化合物25a~d的陰離子配位性質,通過比較發現,化合物25c因為存在1個NH的氫鍵給體,與Cl-的配位能力是化合物25d的95倍,單晶結構顯示Cl-與底部和上部的吡咯同時形成了氫鍵。同時該研究組還發現化合物25c通過與Cl-的緩慢交換過程可以調控DMSO-d6/水體系中Cl-的濃度。Moyer,Sessler,Delmau研究組同時還發現,化合物25a能夠從把溴化銫、氯化銫從水體系提取到硝基苯體系中[59]。在萃取過程中存在一個平衡,氯化銫中氯離子與銫離子的結合能要比溴化銫大7 kJ·mol-1,而吡咯陰離子配合物與銫的自由能對于溴和氯來說則相差不大,所以銫離子可以穩定在吡咯的碗裝空腔內。
Akar,Bielawski,Sessler研究組合成了一類含有杯[4]吡咯的聚合物26b、26c,如圖,通過自由基聚合的方法由單體26a合成了均聚物26b和共聚物26c[60],26b可以從水溶液中萃取四丁基氯化銨和四丁基氟化銨,而26a則沒有這種效果,聚合物不能萃取H2PO4-,這說明與親水性的陰離子相比,疏水性的更容易被萃取。(Cl-:ΔGh=-340 kJ·mol-1;F-:ΔGh=-465 kJ·mol-1;H2PO4-:ΔGh=-465 kJ·mol-1)。

圖14 基于吡咯的陰離子識別主體分子25、26的結構及25c-Cl-的晶體結構Fig.1 4Molecular structure of pyrrole-based anion receptors 25 and 26,and the crystal structures of 25c-Cl-
Sun教授研究組合成了二吡咯的酰胺類化合物27a~e,其中化合物27b、27c表現出對CN-獨特的選擇性[61-62],這是因為發生了親核加成反應,這使得27b、27c可以用作CN-的獨特的傳感器。Denekamp、Suwinska、Eichen研究組合成了三吡咯甲烷的化合物28并研究了其與陰離子配位的性質[63],化合物28表現出對F-獨特的選擇性,其他陰離子,除了H2PO4-,作用都比較弱。化合物28與F-在MeCN-d3中的結合常數為K1=4.1×104mol-1·L、K2=1.7×104mol-1·L,與H2PO4-的結合常數為K1=300 mol-1·L。

圖15 二吡咯-酰胺陰離子識別主體分子27與三吡咯甲烷28的結構及27c識別CN-的機理Fig.1 5Molecular structure of dipyrrole carboxamide 27 and tripyrrolemethane 28,and the sensing mechanism of 25c towards CN-
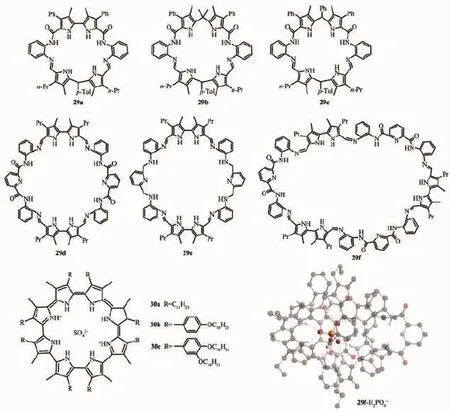
圖16 基于吡咯的大環陰離子識別主體分子29,30的結構及29f-H2PO4-的晶體結構Fig.1 6Molecular structure of pyrrole-based cyclic anion receptors 29 and 30,and the crystal structures of 29f-H2PO4-
Sessler和Katayev教授研究組合成了一類含有酰胺與二吡咯甲烷的環狀陰離子受體分子29a~c[64]。該類化合物表現出與含氧陰離子很強的配位作用,在CH3CN溶液中,通過紫外滴定實驗得到的結合常數達到107的數量級。其中化合物29a與HSO4-作用最強,結合常數達到2.7×106mol-1·L,化合物29b與AcO-配位作用最強,結合常數為4.1× 106mol-1·L,而結構比較剛性的化合物29c則表現出與Cl-比較強的作用力,結合常數為2.81×105mol-1·L。接著該研究組繼續報道了更大的寡聚吡咯的環狀陰離子受體分子29d~f[65-66]。從化合物29f與H2PO4-的單晶結構中可以看出,環29f發生扭曲而與陰離子配位,總共形成了12個氫鍵。通過在CH3CN溶液中的紫外滴定實驗發現,該類化合物表現出對HSO4-、H2PO4-比較好的選擇性。Sessler教授研究組合成了一類環[8]吡咯的衍生物30a~c[67-68],該類化合物有富電子平面,當與缺電子的受體結合時,能形成六角柱狀的液晶相。并且30a可以從溶液中高選擇性的萃取SO42-。

圖17 基于杯吡咯的大環陰離子識別主體分子31、32、33的結構及離子對受體33對Cs+的可控萃取Fig.1 7Molecular structure of calix pyrrole-based cyclic anion receptors 31,32 and 33,and the controlled Cesium recognition by 33
Lee和Sessler研究組報道了一類基于杯吡咯的可以識別手性陰離子的主體分子31(R(+)/S(-))[69],該化合物包含一個具有光學活性的聯萘酚基團,1H NMR研究表明,(S)-31表現出對手性陰離子carboxylate(S)-2-phenylbutyrate((S)-PB)的選擇性識別,與(S)-PB、(R)-PB的結合常數分別為1.0×105mol-1·L、9.8×103mol-1·L。Lee教授研究組合成了一類杯[6]吡咯的化合物32[70],該分子表現出對F-的選擇性,并且有趣的是,表現出一種緩慢的配位/非配位的動力學過程。在加入1倍量F-時,只檢測到對于配合物的共振信號,所以形成作用力很強的配合物。而加入Cl-時,檢測到配位和非配位的吡咯NH的共振信號,比率趨于4∶2。所以6個吡咯中有4個與Cl-形成了氫鍵作用。基于連接有多個陽離子配位位點的陰離子識別杯[4]吡咯的離子對受體33,Sessler及其同事報道其可控識別和陽離子置換性質[71]。核磁研究,氣相優化研究和X射線晶體學研究表明受體與CsCl或CsNO3形成穩定的1∶1配合物。然而,受體和鉀離子間較強的配位意味著添加KClO4會導致從銫離子配位向鉀配位的置換。核磁共振和放射性測量表明受體能夠從重水提取CsCl和CsNO3到硝基苯中。暴露于CHCl3中就可以釋放自由的受體,表明受體可重復使用。
4 基于三氮唑及相關CH…X-氫鍵的陰離子識別主體分子
Flood教授研究組合成了化合物34[72],實現了通過中性的1,2,3-三氮唑的C-H原子與陰離子的氫鍵作用來配位陰離子的方法[73-75]。1H NMR顯示了1,2,3-三氮唑的C-H發生明顯的低場位移,苯環上的C-H的化學位移變化則相對較小,二氯甲烷中的紫外光譜滴定研究顯示34與Cl-的結合常數為1.3×105mol-1·L。改變苯環上的取代基可以調節體系對陰離子的配位能力以及體系的自聚焦能力[76],將三氮唑碳相連的苯環換為吡啶(35),將引起空腔尺寸的改變及分子偶極的改變,從而與碘離子形成三明治夾心結構[77]。同一時期,基于三氮唑構建陰離子識別的折疊體也被報道[78-79]。

圖18 基于三氮唑的陰離子識別主體分子34~43Fig.1 8Molecular structure of triazole-based anion receptors 34~43
我們利用三唑與酰胺的協同作用設計了新型組裝基元-酰胺三唑,并構建了不同形狀、含不同數目酰胺三唑的配體(36~38),發現他們是很好的含氧四面體陰離子識別主體分子[80]。通過[3+2]環加成反應和酸-胺偶聯反應合成了一系列含酰胺三唑的折疊體(39)。利用1H NMR滴定研究了折疊體-陰離子的結合行為和親和力。通過改變酰胺三唑單元的數目以及在終端苯環上引入吸電子取代基來優化折疊體的結構,以便于高度選擇性地識別硫酸根陰離子。結果表明受體和陰離子之間的結構相容性對高選擇性陰離子結合,以及電子效應對調節受體的陰離子結合力很關鍵[81]。將酰胺三唑單元與分子內電荷轉移生色團結合,獲得了一種包含OH,NH和CH 3種氫鍵給體的陰離子受體40,通過氟離子對分子內電荷轉移的調控實現了對氟離子高效的“裸眼”檢測[82]。將酰胺三唑與手性單元結合,我們還獲得了對硫酸根陰離子具有手性信號相應的大環分子41[83]。通過三氮唑將2個卟啉單元連接起來構筑了卟啉籠狀化合物42,利用卟啉鋅原子與氮的軸向配位實現了對疊氮陰離子的高效識別[84]。我們合成了一個由光活性的卟啉單元及吡啶單元與剛性的聯苯連接單元組成的大環分子43,將該大環分子中的吡啶與三氮唑甲基化后得到對陰離子有很好的配位能力的大環主體分子。利用該大環分子的納米空腔及其對陰離子的配位能力,研究了它對TCNQ陰離子客體分子二聚反應的催化[85]。
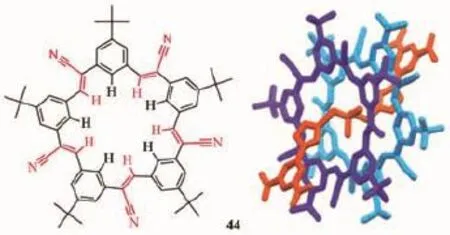
圖19 五角形大環分子44的結構及其所形成的[3]輪烷Fig.1 9Molecular structure of cyanostar 44,and the[3] rotaxane based on it
Flood教授還設計了五角形大環分子“cyanostar”44以配位六氟磷酸根等大陰離子,其以大環/陰離子2∶1的方式配位。該大環還可以結合二烷基磷酸鹽分子軸形成[3]輪烷[86]。
5 基于銨鹽的陰離子識別主體分子
最早的合成陰離子受體就是通過質子化的銨鹽來實現的,如今這項研究仍在繼續,含銨鹽的陰離子識別主體分子即使在水體系中仍能表現出對于陰離子良好的識別。
Felix教授研究組合成了化合物45[87],該化合物包含兩個鄰菲羅啉基團,電化學和1H NMR研究說明該化合物可以用來識別1,3,5-三苯甲酸鹽(BTC3-)和芘甲酸鹽(PYRC-)。pH值分別為5.5和3.0。實驗和模擬研究都表明通過+N-H…-O的氫鍵作用以及鄰菲羅啉與客體的π-π堆積作用,化合物45能夠與陰離子客體配位在一起。
Martinez和Llobet教授研究組合成了環46以及甲基化后的Me246[88],并對兩種化合物配位陰離子的性質做了比較。發現Me246對對苯二甲酸有一定的選擇性,而46表現出對鄰苯二甲酸良好的選擇性。這歸因于在46中,中間的二級胺與甲基化的Me246相比,轉動的程度要小,而且存在氫鍵作用。
Nelissen和Smith研究組合成了開環的47a,環狀的化合物47b,其在水溶液中表現出對于PO4-的強的配位作用[89]。通過滴定含有PO4-的熒光化合物47c確定了47a、47b對于PO4-的親和力,lgK分別為5.15和5.30。電化學研究表明在大量Cl-存在下,47a、47b仍然與無機的PO4-有很強的作用,lgK分別為4.4~8.6和7.5~14.2,最大的結合常數是通過胺基質子化后得到的,也說明了靜電作用力起了重要作用。

圖20 基于銨鹽的大環陰離子識別主體分子45、46和47的結構Fig.2 0Molecular structure of ammonium-based cyclic anion receptors 45,46 and 47
6 基于胍鹽的陰離子識別主體分子
胍鹽也是一類配位含氧陰離子的良好的受體分子[90-91],主要通過靜電作用力和氫鍵來配位陰離子。與銨鹽相比,該類化合物可以在比較寬的pH范圍內保持質子化的狀態。Lehn教授研究組在上世紀70年代末報道了第一例通過胍鹽基團來識別PO32-的環狀識別主體。48a、48b、48c在水溶液中與PO32-的結合常數分別為50、158、251 mol-1·L。
Mendoza教授研究組合成了化合物49a[91],其中對離子為Cl-,該化合物與對硝基苯甲酸的結合常數只有1.6×103mol-1·L。對離子在配位過程中起著很重要的作用,當把Cl-換成結合力較弱的六氟磷酸根或者四苯基硼酸根時,結合常數明顯變大。1H NMR的研究表明對硝基苯甲酸根負離子與NH形成強的氫鍵作用,同時,萘與硝基苯甲酸根之間還存在π-π堆積作用。該研究組還合成了非環化的基于胍鹽的49b和環化的49c~e并比較了它們與NO3-的配位能力[92],配位常數分別為5.38×103、73.7× 103mol-1·L,結果發現,環化分子的配位能力要強的多。
7 基于咪唑的陰離子識別主體分子
咪唑中酸化的CH能夠很好的配位陰離子,Alcalde研究組通過晶體結構證實了化合物50[93-94]中這類氫鍵的存在[95]。Hwang和Kim教授研究組合成了一系列基于咪唑與喹啉的陰離子識別主體分子51a~d[95-96],并通過熒光光譜研究了它們與陰離子作用。游等[97]發現由2個剛性的四咪唑鎓大環分子52協同能極大增強分子識別,在水相中獲得了高達8.6×109mol-2·L2的硫酸根離子高選擇性熒光識別。除靜電作用外,X-射線單晶衍射分析表明,硫酸根是通過八個咪唑鎓單元的C2氫原子和硫酸鹽的氧原子之間的氫鍵包裹在由正交排列的2個大環所形成的三明治夾心結構的假六面體空腔中。這個三明治結構由苯基和三嗪酮環之間的π-π堆積及多個外圍鏈和剛性骨架之間的電荷輔助的氫鍵所增強。這些外圍-骨架間氫鍵能提供柔性的外鏈側鏈以包裹三明治結構,以防止硫酸根被水所溶劑化。這個結合過程伴隨著熒光增強,這歸因于旋轉受限以及配位硫酸根后大環骨架更好的π共軛。

圖21 基于胍鹽的陰離子識別主體分子48、49的結構Fig.2 1Molecular structure of guanidinium-based cyclic anion receptors 48 and 49

圖22 基于咪唑的大環陰離子識別主體分子50、51和52的結構Fig.2 2Molecular structure of immidazole-based cyclic anion receptors 50,51 and 52
8 基于羥基的陰離子識別主體分子
近年來,羥基作為一種陰離子的識別單元也被廣泛的研究,Smith教授研究組在把鄰苯二酚用作陰離子受體的研究上做了大量的工作。合成了一系列基于鄰苯二酚的陰離子識受體分子53a~j[98],分子內氫鍵在配位陰離子過程中起了關鍵作用,在化合物53a~c中,酰胺的氧原子與羥基形成氫鍵,羥基之間也存在氫鍵,能夠與氯離子配位的位點只剩下酰胺,53d、53e中存在類似的分子內氫鍵,所以與氯離子沒有作用。通過一系列的比較試驗,發現53j對于氯離子配位能力最好。

圖23 基于羥基的陰離子識別主體分子53的結構及與陰離子作用力大小比較Fig.2 3Molecular structure of hydroxyl-based anion receptors 53 and the binding affinity comparison
9 結論與展望
總之,自從陰離子識別的超分子化學發展起來以來,大量的陰離子識別主體分子被設計合成出來,它們含有不同的識別單元,通過各種非共價鍵的作用力與不種陰離子配位。當然,優秀的識別主體往往包含著若干個識別單元,充分利用這些識別單元來構建新穎的陰離子識別主體分子成為研究的首要任務。同時,將各種電子、質子、能量轉移過程(例如光致電子/能量轉移,基態/激發態分子內/分子間質子轉移,光誘導電荷轉移)巧妙地與各種識別主體分子結合起來,實現對陰離子的識別、對體系的光物理性能的調控,模擬生物體系的離子傳輸[99-101]也將是關注的重點。
[1]Park C H,Simmons H E.J.Am.Chem.Soc.,1968,90:2431-2432
[2]Pedersen C J.J.Am.Chem.Soc.,1967,89:7017-7036
[3]Gale P A,Perez-Tomas R,Quesada R.Acc.Chem.Res.,2013, 46:2801-2813
[4]Davis J T,Okunola O,Quesada R.Chem.Soc.Rev.,2010, 39:3843-3862
[5]Davis A P,Sheppard D N,Smith B D.Chem.Soc.Rev., 2007,36:348-357
[6]Steed J W.Chem.Soc.Rev.,2010,39:3686-3699
[7]Evans N H,Beer P D.Angew.Chem.Int.Ed.,2014,53:11716 -11754
[8]Gale P A,Busschaert N,Haynes C J E,et al.Chem.Soc. Rev.,2014,43:205-241
[9]Pascal R A,Spergel J,Vanengen D.Tetrahedron Lett.,1986, 27:4099-4102
[10]Kavallieratos K,Bertao C M,Crabtree R H.J.Org.Chem., 1999,64:1675-1683
[11]Sambrook M R,Beer P D,Wisner J A,et al.J.Am.Chem. Soc.,2005,127:2292-2302
[12]Hughes M P,Smith B D.J.Org.Chem.,1997,62:4492-4499
[13]Santacroce P V,Okunola O A,Zavalij P Y,et al.Chem. Commun.,2006:3246-3248
[14]Kang S O,Begum R A,Bowman-James K.Angew.Chem.Int.Ed.,2006,45:7882-7894
[15]Rodriguez-Docampo Z,Pascu S I,Kubik S,et al.J.Am. Chem.Soc.,2006,128:11206-11210
[16]Reyheller C,Kubik S.Org.Lett.,2007,9:5271-5274
[17]Chmielewski M J,Jurczak J.Chem.-Eur.J.,2005,11:6080-6094
[18]Chmielewski M J,Jurczak J.Chem.-Eur.J.,2006,12:7652-7667
[19]Chmielewski M J,Zielinski T,Jurczak J.Pure Appl.Chem., 2007,79:1087-1096
[20]Shang X F,Xu X F,Lin H,et al.J.Inclusion Phenom. Macrocyclic Chem.,2007,58:275-281
[21]Shao J,Lin H,Shang X F,et al.J.Inclusion Phenom. Macrocyclic Chem.,2007,59:371-375
[22]Hossain M A,Llinares J M,Powell D,et al.Inorg.Chem., 2001,40:2936-2937
[23]Hossain M A,Kang S O,Powell D,et al.Inorg.Chem.,2003, 42:1397-1399
[24]Ghosh S,Roehm B,Begum R A,et al.Inorg.Chem.,2007, 46:9519-9521
[25]Kim S K,Bok J H,Bartsch R A,et al.Org.Lett.,2005,7: 4839-4842
[26]Choi J K,Kim S H,Yoon J,et al.J.Org.Chem.,2006,71: 8011-8015
[27]Kang S O,Day V W,Bowman-James K.Org.Lett.,2008,10: 2677-2680
[28]Beeren S R,Sanders J K M.J.Am.Chem.Soc.,2011,133: 3804-3807
[29]Edwards S J,Valkenier H,Busschaert N,et al.Angew.Chem. Int.Ed.,2015,54:4592-4596
[30]Wu Y,Peng X,Fan J,et al.J.Org.Chem.,2006,72:62-70
[31]Burns D H,Calderon-Kawasaki K,Kularatne S.J.Org. Chem.,2005,70:2803-2807
[32]Calderon-Kawasaki K,Kularatne S,Li Y H,et al.J.Org. Chem.,2007,72:9081-9087
[33]Lakshminarayanan P S,Ravikumar I,Suresh E,et al.Chem. Commun.,2007:5214-5216
[34]Jose D A,Kumar D K,Ganguly B,et al.Inorg.Chem.,2007, 46:5817-5819
[35]Dahan A,Ashkenazi T,Kuznetsov V,et al.J.Org.Chem., 2007,72:2289-2296
[36]Brooks S J,García-Garrido S E,Light M E,et al.Chem.-Eur.J.,2007,13:3320-3329
[37]Brooks S J,Gale P A,Light M E.Chem.Commun.,2006: 4344-4346
[38]Meshcheryakov D,Bhmer V,Bolte M,et al.Chem.-Eur.J., 2009,15:4811-4821
[39]Meshcheryakov D,Arnaud-Neu F,Bohmer V,et al.Org. Biomol.Chem.,2008,6:1004-1014
[40]Meshcheryakov D,Arnaud-Neu F,Bohmer V,et al.Org. Biomol.Chem.,2008,6:3244-3255
[41]Custelcean R,Bonnesen P V,Duncan N C,et al.J.Am. Chem.Soc.,2012,134:8525-8534
[42]Jia C,Wu B,Li S,et al.Angew.Chem.Int.Ed.,2011,50: 486-490
[43]Wezenberg S J,Vlatkovic M,Kistemaker J C M,et al.J. Am.Chem.Soc.,2014,136:16784-16787
[44]Sessler J L,Camiolo S,Gale P A.Coord.Chem.Rev.,2003, 240:17-55
[45]Gale P A.Chem.Commun.,2008:4525-4540
[46]Curiel D,Cowley A,Beer P D.Chem.Commun.,2005:236-238
[47]Chmielewski M J,Zhao L,Brown A,et al.Chem.Commun., 2008:3154-3156
[48]Chang K J,Moon D,Lah M S,et al.Angew.Chem.Int.Ed., 2005,44:7926-7929
[49]Kim N K,Chang K J,Moon D,et al.Chem.Commun.,2007: 3401-3403
[50]Kwon T H,Jeong K S.Tetrahedron Lett.,2006,47:8539-8541
[51]Ju J,Park M,Suk J m,et al.Chem.Commun.,2008:3546-3548
[52]Zhao Y,Li Y,Li Y,et al.Org.Biomol.Chem.,2010,8:3923 -3927
[53]Bates G W,Gale P A,Light M E.Chem.Commun.,2007: 2121-2123
[54]Caltagirone C,Hiscock J R,Hursthouse M B,et al.Chem.-Eur.J.,2008,14:10236-10243
[55]Caltagirone C,Gale P A,Hiscock J R,et al.Chem.Commun., 2008:3007-3009
[56]Gale P A,Hiscock J R,Moore S J,et al.Chem.-Asian J., 2010,5:555-561
[57]Yoon D W,Gross D E,Lynch V M,et al.Angew.Chem. Int.Ed.,2008,47:5038-5042
[58]Yoon D W,Gross D E,Lynch V M,et al.Chem.Commun., 2009:1109-1111
[59]Wintergerst M P,Levitskaia T G,Moyer B A,et al.J.Am. Chem.Soc.,2008,130:4129-4139
[60]Aydogan A,Coady D J,Lynch V M,et al.Chem.Commun., 2008:1455-1457
[61]Chen C L,Lin T P,Chen Y S,et al.Eur.J.Org.Chem., 2007:3999-4010
[62]Chen C L,Chen Y H,Chen C Y,et al.Org.Lett.,2006,8: 5053-5056
[63]Denekamp C,Suwinska K,Salman H,et al.Chem.-Eur.J., 2007,13:657-665
[64]Katayev E A,Boev N V,Khrustalev V N,et al.J.Org.Chem.,2007,72:2886-2896
[65]Katayev E A,Sessler J L,Khrustalev V N,et al.J.Org. Chem.,2007,72:7244-7252
[66]Katayev E A,Pantos G D,Reshetova M D,et al.Angew. Chem.Int.Ed.,2005,44:7386-7390
[67]Seidel D,Lynch V,Sessler J L.Angew.Chem.Int.Ed., 2002,41:1422-1425
[68]Stpień M,Donnio B,Sessler J L.Angew.Chem.Int.Ed., 2007,46:1431-1435
[69]Miyaji H,Hong S J,Jeong S D,et al.Angew.Chem.Int. Ed.,2007,46:2508-2511
[70]Yoon D W,Jeong S D,Song M Y,et al.Supramol.Chem., 2007,19:265-270
[71]Kim S K,Vargas-Zuniga G I,Hay B P,et al.J.Am.Chem. Soc.,2012,134:1782-1792
[72]Li Y J,Flood A H.Angew.Chem.Int.Ed.,2008,47:2649-2652
[73]Hua Y,Flood A H.Chem.Soc.Rev.,2010,39:1262-71
[74]Li Y,Griend D A V,Flood A H.Supramol.Chem.,2009, 21:111-117
[75]Zahran E M,Hua Y,Li Y,et al.Anal.Chem.,2010,82:368-375
[76]Li Y,Flood A H.J.Am.Chem.Soc.,2008,130:12111-12122
[77]Li Y,Pink M,Karty J A,et al.J.Am.Chem.Soc.,2008, 130:17293-17295
[78]Juwarker H,Lenhardt J M,Pham D M,et al.Angew.Chem. Int.Ed.,2008,47:3740-3743
[79]Hua Y,Flood A H.J.Am.Chem.Soc.,2010,132:12838-12840
[80]Li Y J,Xu L,Yang W L,et al.Chem.-Eur.J.,2012,18:4782 -4790
[81]Cao L,Jiang R,Zhu Y,et al.Eur.J.Org.Chem.,2014:2687-2693
[82]Xu L,Li Y,Yu Y,et al.Org.Biomol.Chem.,2012,10:4375 -4380
[83]Jiang R,Li Y,Qin Z,et al.RSC Adv.,2014,4:2023-2028
[84]Zhang J,Li Y,Yang W,et al.Chem.Commun.,2012,48: 3602-3604
[85]Li Y J,Zhao Y J,Flood A H,et al.Chem.-Eur.J.,2011, 17:7499-7505
[86]Lee S,Chen C H,Flood A H.Nat.Chem.,2013,5:704-710
[87]Cruz C,Delgado R,Drew M G B,et al.J.Org.Chem.,2007, 72:4023-4034
[88]Arbuse A,Anda C,Martínez M A,et al.Inorg.Chem.,2007, 46:10632-10638
[89]Nelissen H F M,Smith D K.Chem.Commun.,2007:3039-3041
[90]Blondeau P,Segura M,Perez-Fernandez R,et al.Chem. Soc.Rev.,2007,36:198-210
[91]Echavarren A,Galan A,Lehn J M,et al.J.Am.Chem.Soc., 1989,111:4994-4995
[92]Blondeau P,Benet-Buchholz J,de Mendoza J.New J.Chem., 2007,31:736-740
[93]AlcaldeE,MesquidaN,Perez-GarciaL,etal.Chem.Commun., 1999:295-296
[94]Yoon J,Kim S K,Singh N J,et al.Chem.Soc.Rev.,2006, 35:355-360
[95]Chellappan K,Singh N J,Hwang I C,et al.Angew.Chem. Int.Ed.,2005,44:2899-2903
[96]Singh N J,Jun E J,Chellappan K,et al.Org.Lett.,2007,9: 485-488
[97]Zhou H,Zhao Y,Gao G,et al.J.Am.Chem.Soc.,2013, 135:14908-14911
[98]Winstanley K J,Smith D K.J.Org.Chem.,2007,72:2803-2815
[99]Ko S K,Kim S K,Share A,et al.Nat.Chem.,2014,6:885-892
[100]Parker J L,Newstead S.Nature,2014,507:68-72
[101]Sun J,Bankston J R,Payandeh J,et al.Nature,2014,507: 73-77
Recent Development of Anion Receptors Based on Hydrogen Bonding
LI Yong-Jun*LIU Hui-BiaoLI Yu-Liang
(CAS Key Laboratory of Organic Solids,Institute of Chemistry,Chinese Academy of Sciences,Beijing 100190,China)
The important roles of anions in biology,medicine,catalysis and environmental science have been widely recognized.Anion receptors showed bright application prospects in the trans epithelial ion transportation, chemical sensing,simulation of enzyme catalyzed organic reactions.Here we summarize the recent progress of anion receptors based on different hydrogen bonding units such as amide,urea/thiourea,indole/pyrroles,triazole, ammonium,guanidinium,imidazole,hydroxyl groups.
anion recognization;anion receptors;hydrogen bonding
O641.3;O641.4
A
1001-4861(2015)09-1687-18
10.11862/CJIC.2015.252
2015-05-27。收修改稿日期:2015-07-19。
國家自然科學基金(No.21290190,21322301)資助項目。
*通訊聯系人。E-mail:liyj@iccas.ac.cn
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
南大法學(2021年3期)2021-08-13 09:22:32
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
阿來研究(2021年1期)2021-07-31 07:39:04
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
中國自行車(2018年9期)2018-10-13 06:17:10
汽車工程學報(2017年2期)2017-07-05 08:13:02
金色年華(2016年13期)2016-02-28 01:43:27
