正交試驗法優選葛根配方顆粒的提取工藝
2015-12-14 05:08:24杜遠東
中國民族民間醫藥 2015年15期
關鍵詞:工藝
杜遠東
陜西省漢中市食品藥品檢驗檢測中心,陜西 漢中 723000
葛根為豆科植物野葛Pueraria lobata (Wild.)Ohwi 或甘葛藤P.thomsonii Benth 的干燥根,其主要活性成分為異黃酮類化合物,如葛根素、大豆素、大豆苷等。葛根配方顆粒是葛根飲片經水提、濃縮、干燥、制粒等工序制成的單味中藥顆粒劑[1]。將葛根飲片制備為顆粒劑,可以有效地去除植物纖維等無活性成分,增加穩定性,易儲存,服用方便。
1 材料與儀器
1.1 材料 葛根(西安盛興飲片廠);葛根素對照品(批號:0766-200611,中國藥品生物制品檢定所);甲醇(色譜級);超純水(實驗室自制)。
1.2 儀器 Waters 高效液相色譜儀(Waters2695 Separations Module,Waters2996 PAD 檢測器),含在線真空脫氣機、四元梯度泵、自動進樣器、柱溫箱;梅特勒GB204 電子天平;SB3200DT 超聲波清洗機(寧波新莖生物科技股份有限公司);101 型電熱鼓風干燥箱(北京科偉永鑫實驗儀器設備廠);艾柯-Advanced-06 型超純水機(成都唐氏康寧科技發展有限公司)。
2 方法與結果
2.1 提取方法
2.1.1 采用傳統工藝提取 稱取100g 藥材,加入水量沒過藥材面1~2cm 即可,浸泡0.5h,用武火煎煮至沸騰,然后改用文火煎煮,30min/次,提取2 次,合并藥液,濃縮,定容至250ml,備用。
2.1.2 水沸后提取 稱取100g 藥材,加入沸水中,水量以沒過藥材面1~2cm 即可,武火煮沸后,改用文火,30min/次,提取2 次,合并藥液,濃縮,定容至250ml,備用。
2.1.3 水提 稱取藥材100g,分別加入10、8 倍量的水提取,第一次提取1h,第二次提取30min,濾出藥液,合并藥液,濃縮,定容至250ml,備用。
2.1.4 60%乙醇提取[2]稱取100g 藥材,加入5 倍量的60%乙醇回流提取,第一次提取1.5h,第二次提取1 h,濾取藥液,合并藥液,減壓回收乙醇,定容至250ml,備用。
2.1.5 95% 乙醇提取 稱取100g 藥材,加入5 倍量的95%乙醇回流提取,第一次提取1.5h,第二次提取1 h,濾取藥液,合并藥液,減壓回收乙醇,定容至250ml,備用。
2.2 高效液相色譜測定含量
2.2.1 色譜條件[3]以十八烷基硅烷鍵合硅膠為填充劑;流動相:甲醇-水(25∶75);檢測波長:250nm。
2.2.2 對照品溶液的制備 精密稱取葛根素標準品10.4mg,加30%的乙醇定容至50ml,稀釋5 倍,制成2mg/50ml 的對照品溶液,用微孔濾膜過濾,備用。
2.2.3 供試品溶液 吸取各樣品溶液2ml,用30%的乙醇定容至100ml,用微孔濾膜過濾,備用。
2.2.4 結果 由表1 可知,60%乙醇提取所得的葛根素含量最高,因此溶劑選擇60%乙醇。

表1 不同提取方法葛根素含量結果
2.3 正交設計試驗優選葛根提取工藝 選取浸泡時間(A)、加醇量(B)、提取時間(C)、提取次數(D)四個水平,以葛根素含量與干膏收率為指標的進行正交試驗,優化葛根提取工藝。按“2.2”的方法測定葛根素含量,因素水平、正交試驗、正交試驗結果、直觀分析、方差分析的結果見表2、3、4、5、6。

表2 正交因素水平表
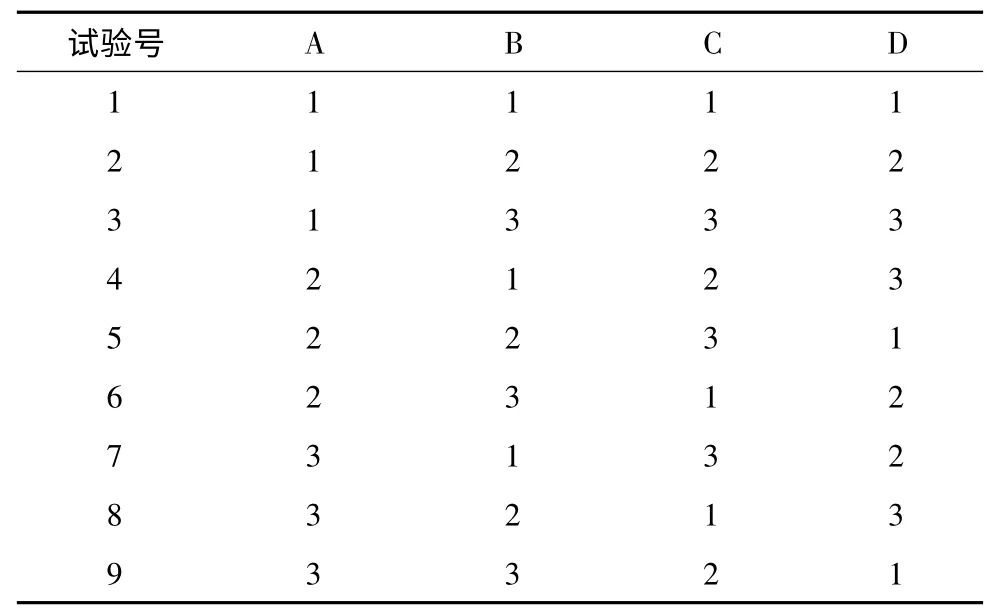
表3 正交試驗設計
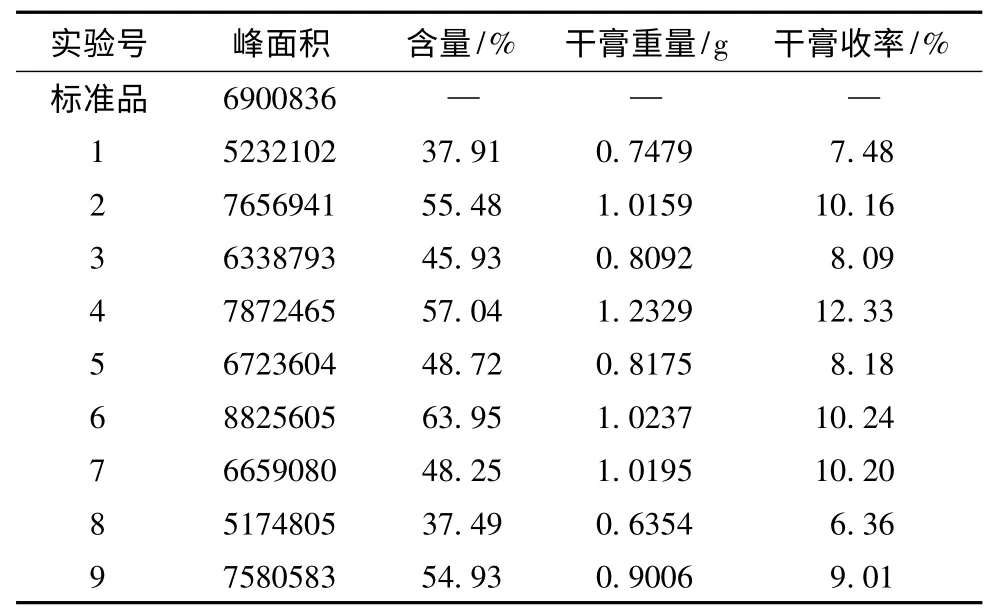
表4 正交試驗結果

表5 直觀分析

表6 方差分析
以干膏收率與葛根素含量的綜合評分為考察指標,直觀分析表(表5)中極差R 值最小的為誤差項。對各因素進行直觀分析,A 因素:K2>K3>K1;C 因素:K2>K3>K1。D 因素:K2>K3>K1;B 因素干膏收率為K1>K3>K2;含量測定為K2>K3>K1,綜合考慮,B 因素選K3。根據以上結果分析得出工藝為A2B3C2D2,即為用60%乙醇,12倍的量,浸泡1h,提取2 次,每次1.5 h。
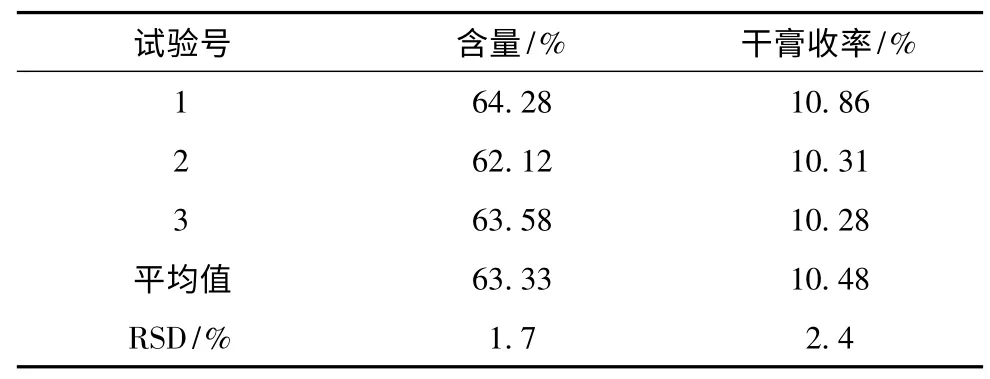
表7 驗證試驗結果
2.4 驗證試驗 分別稱取20g 葛根藥材,按優選最佳工藝條件進行3 次驗證試驗,結果見表7。
3 結論
正交試驗結果表明,該優選工藝合理、穩定、重現性好,為葛根配方顆粒的生產工藝奠定了基礎。
[1]顧志平,陳碧珠,馮瑞之.中藥葛根及其同屬植物資源的利用和評價[J].藥學學報,1996,31 (5):3.
[2]匡海學.中藥化學[M].1 版.北京:中國中醫藥出版社,2003:178.
[3]國家藥典委員會.中華人民共和國藥典(第一部)[S].北京:中國醫藥科技出版社,2010:313.
猜你喜歡
中國特種設備安全(2022年5期)2022-08-26 09:19:32
礦產綜合利用(2020年1期)2020-07-24 08:50:40
山東冶金(2019年6期)2020-01-06 07:45:54
收藏界(2019年2期)2019-10-12 08:26:06
世界農藥(2019年2期)2019-07-13 05:55:12
世界農藥(2019年2期)2019-07-13 05:55:10
模具制造(2019年3期)2019-06-06 02:11:00
山東工業技術(2016年15期)2016-12-01 05:30:59
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
