體外高糖環境對H9C2心肌細胞增殖的影響及其機制探討
2016-01-20 13:55:30盧海龍楊榮禮李雷徐州醫學院附屬醫院江蘇徐州221006
山東醫藥 2015年28期
盧海龍,楊榮禮,李雷(徐州醫學院附屬醫院,江蘇徐州221006)
體外高糖環境對H9C2心肌細胞增殖的影響及其機制探討
盧海龍,楊榮禮,李雷
(徐州醫學院附屬醫院,江蘇徐州221006)
摘要:目的觀察體外高糖環境對H9C2心肌細胞增殖的影響,并探討其作用機制。方法將體外培養鼠胚心肌細胞H9C2隨機分為四組,1組培養液中加入終濃度為5.5 mmol/L的D-葡萄糖,2組加入終濃度為27.5 mmol/L的甘露醇和5.5 mmol/L的D-葡萄糖,3組加入終濃度為33 mmol/L的D-葡萄糖,4組加入終濃度為2 μmol/L的經典瞬時受體蛋白( TRPC)阻滯劑SKF96365和終濃度為33 mmol/L的D-葡萄糖。分別在培養24、48、72 h時,采用CCK8法測定細胞增殖率,實時定量PCR法測定經典瞬時受體蛋白6( TRPC6)、活化T細胞核因子3( NFAT3) mRNA; 72 h時采用Western blotting法檢測TRPC6及NFAT3蛋白。結果培養72 h時1、2、3、4組細胞增殖率分別為105.88%±10.01%、98.67%±8.97%、28.55%±6.32%、49.18%±7.14%,3組較1、2組降低( P均<0.05),4組較3組增高( P<0.05) ; 3組TRPC6、NFAT3 mRNA及蛋白相對表達量較1、2組增加( P均<0.05),4組較3組下降( P均<0.05)。結論高糖抑制心肌細胞增殖;促進心肌細胞TRPC6表達,導致TRPC通道開放,啟動鈣調神經素( CaN)/活化T細胞核因子( NFAT)通路,可能是其作用機制。
關鍵詞:糖尿病心肌病;高糖;鼠胚心肌細胞株H9C2;經典瞬時受體蛋白6;活化T細胞核因子3
糖尿病心肌病( DCM)早期表現為舒張性左心功能不全,晚期容易并發惡性心律失常、心力衰竭、甚至猝死[1,2]。迄今為止,DCM發病機制仍不完全明確,治療未有實質性突破[3]。研究發現,心肌細胞內鈣超載與DCM的發生有關[4]。高血糖是糖尿病患者的主要特征之一,能夠直接損傷心肌細胞,引起心肌病變[5]。但是,高血糖對心肌細胞內鈣超載是否有促進作用及其可能機制,文獻報道較少。2012年9月~2013年7月,本研究以體外培養的鼠胚心肌細胞H9C2為研究對象,觀察體外高糖環境對細胞形態及增殖的影響,并探討其作用機制。
1 材料與方法
1.1材料鼠胚心肌細胞H9C2購自中國科學院上海生命科學研究院。DMEM培養基、D-葡萄糖、胎牛血清( FBS) (美國Gibco公司),細胞計數盒CCK-8(日本Dojindo Lab),細胞總RNA提取試劑盒、核蛋白提取試劑盒、經典瞬時受體蛋白6( TRPC6)和GAPDH引物、DNA marker(上海生物工程有限公司),實時定量PCR試劑盒(大連TaKaRa公司),兔抗大鼠TRPC6單克隆抗體、兔抗大鼠活化T細胞核因子3 ( NFAT3)單克隆抗體、經典瞬時受體通道蛋白( TRPC)通道阻滯劑SKF96365(以色列Alomone Labs公司)。
1.2細胞培養及分組處理H9C2細胞在含10% FBS的低糖( 5 mmol/L) DMEM培養基中培養,待細胞生長85%左右培養面積后,0.25%胰酶消化,1∶2傳代,8~9代細胞用于實驗。待細胞成對數生長后,調整細胞數為1×105/L,接種于6孔培養板,培養24 h;待細胞成融合狀態換用無血清低糖DMEM培養基,繼續培養24 h,使細胞呈靜止狀態,隨機分為四組。1組:培養液中加入終濃度為5.5 mmol/L 的D-葡萄糖; 2組:培養液中加入終濃度為27.5 mmol/L的甘露醇和終濃度為5.5 mmol/L的D-葡萄糖; 3組:培養液中加入終濃度為33 mmol/L的D-葡萄糖; 4組:培養液中加入終濃度為2 μmol/L的SKF96365和終濃度為33 mmol/L的D-葡萄糖。
1.3細胞存活率檢測將各組細胞接種于96孔培養板,分別在培養24、48、72 h時每孔加入CCK-8 10 μL和DMEM 90 μL,37℃、5% CO2條件下孵育1 h,采用酶標儀(λ= 450 nm)記錄各孔光密度( OD)值,計算細胞增殖率。細胞增殖率( %) = (處理組OD450-空白孔OD450)/(對照組OD450-空白孔
OD450)×100%,每組設3個復孔,實驗重復3次。空白孔為未加細胞懸液的DMEM培養液,1組即對照組。
1.4 TRPC6、NFAT3 mRNA檢測采用實時定量PCR方法。各組細胞分別在處理24、48、72 h提取細胞總RNA,測定RNA濃度。TRPC6引物序列:上游: 5'-GTCGGTGGTCATCAACTACAATC-3',下游: 5'-CCACATCCGCATCATCCTCAATT-3'; NFAT3引物序列:上游: 5'-ACAGAAGAGGGGGCGTCTC-3',下游: 5'-ACCAATAGGGGCAGCACCATA-3'; GAPDH引物序列:上游: 5'-ACGGCAAATTCAACGGCACAGTCA-3',下游: 5'-TGGGGGCATCGGCAGAAGG-3'。PCR反應條件: 95℃變性5 min、95℃30 s、58℃30 s、70℃30 s,40個循環。實驗重復3次。以GAPDH為內參照,2-ΔΔCt法計算各組TRPC6、NFAT3 mRNA相對表達量。
1.5 TRPC6、NFAT3蛋白檢測各組細胞在培養72 h時(根據前期實驗結果確定時間點)采用0.25%胰酶常規消化并收集,采用Western blotting法檢測TRPC6( 1∶500)、NFAT3( 1∶2 000)蛋白。具體步驟參照試劑盒說明書。實驗重復3次,以GAPDH作為內參照。采用Quantity-One軟件分析各組TRPC6、NFAT3蛋白的相對表達量。
1.6統計學方法采用SPSS13.0統計軟件。符合正態分布的計量資料采用珋x±s表示,組間比較采用單因素方差分析,兩兩比較采用LSD-t檢驗。P<0.05為差異有統計學意義。
2 結果
2.1各組細胞增殖率比較在培養72 h時,3組細胞增殖率較1、2組降低( P均<0.05),4組較3組增高( P<0.05) ; 3組72 h時細胞增殖率較48 h時降低( P<0.05) ; 1、2組細胞增殖率在各時間點均無明顯差異( P均>0.05) ;見表1。
表1 四組不同時間點細胞增殖率比較( %,±s)
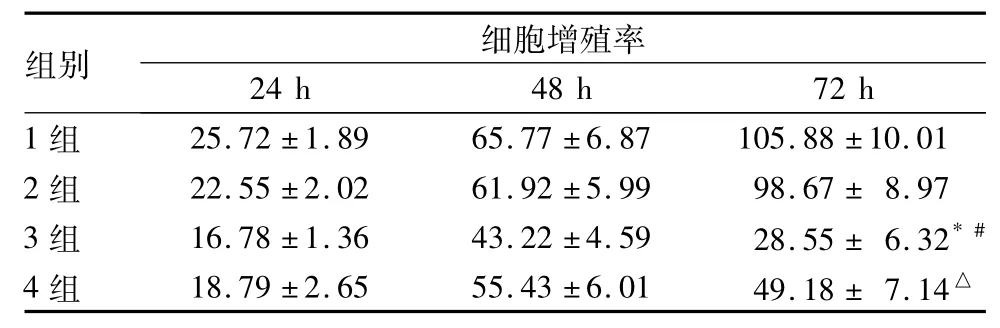
表1 四組不同時間點細胞增殖率比較( %,±s)
注:與同時間點1、2組比較,*P均<0.05;與同時間點3組比較,△P<0.05;與同組48 h比較,#P<0.05。
組別 細胞增殖率24 h 48 h 72 h 1組25.72±1.89 65.77±6.87 105.88±10.01 2組 22.55±2.02 61.92±5.99 98.67±8.97 3組 16.78±1.36 43.22±4.59 28.55±6.32* #4組 18.79±2.65 55.43±6.01 49.18±7.14△
2.2各組TRPC6、NFAT3 mRNA相對表達量比較
在培養72 h時,3組TRPC6、NFAT3 mRNA相對表達量較1、2組增加( P均<0.05),4組較3組下降( P均<0.05) ; 3組各時間點比較,P均<0.05;見表2。
表2 四組不同時間點TRPC6、NFAT3 mRNA相對表達量比較(±s)

表2 四組不同時間點TRPC6、NFAT3 mRNA相對表達量比較(±s)
注:與同時間點1、2組比較,*P均<0.05;與同時間點3組比較,△P<0.05;與同組各時間點比較,#P<0.05。
組別TRPC6 mRNA 24 h 48 h 72 h NFAT3 mRNA 24 h 48 h 72 h 1組 1.03±0.13 0.96±0.09 1.02±0.11 0.97±0.17 0.99±0.13 1.06±0.19 2組 1.07±0.08 1.12±0.15 1.09±0.26 1.14±0.14 1.11±0.11 1.13±0.18 3組 7.23±0.52 11.39±1.17 16.15±1.49* # 5.23±1.43 9.88±1.96 14.44±2.09* #4組 4.64±0.35 6.79±0.44 10.13±0.69# 2.94±0.24 5.32±0.94 8.97±1.73△
2.3各組TRPC6、NFAT3蛋白相對表達量比較在培養72 h時,1、2、3、4組TRPC6蛋白相對表達量分別為0.302±0.017、0.351±0.023、1.874± 0.124、1.214±0.097; NFAT3蛋白相對表達量分別為0.247±0.026、0.298±0.034、1.199±0.104、0.656±0.038; 3、4組均高于1、2組( P均<0.05),4組均低于3組( P均<0.05)。
3 討論
研究發現,持續高血糖狀態會造成心肌細胞胞質內、線粒體內Ca2 +濃度明顯升高,導致心肌細胞舒縮功能障礙及凋亡增加[5,6]。在病理狀態下,胞外鈣的內流和胞內鈣庫的釋放則造成細胞質內鈣超載[7]。細胞外鈣內流主要通過電壓門控鈣信道( VOC)、受體門控鈣通道( ROC)和鈣庫操縱性鈣通道( SOC),其中VOC和ROC主要介導胞外鈣在短時間內大量內流,SOC則介導小量持續鈣內流。但是,組成SOC及ROC的具體分子實體仍不十分明確。研究發現,TRPC的同源異構體是構成細胞膜上SOC和ROC的分子實體,TRPC通道的啟動開放可引起心肌細胞內Ca2 +濃度變化,通過鈣調神經素( CaN)/NFAT信號通路在細胞內Ca2 +增加誘導的心肌重構過程中發揮關鍵作用,進而觸發心肌肥大相關信號轉導,引起心肌重構和病理性肥厚[8]。Onohara等[9]發現,血管緊張素Ⅱ( AngⅡ)誘導的新生大鼠心肌細胞肥大模型TRPC3、TRPC6 mRNA表達均顯著上調;而敲除TRPC3 或TRPC6中的任何一個,都會明顯抑制心肌細胞肥大效應; PLC介導的1,2-二酰甘油( DAG)的生成增多對上述過程有直接啟動作用。TRPC3與TPRC6均可直接受DAG的啟動開放,TRPC3與TRPC6通道之間有協同關系。研究發現,TRPC6表達程度與心肌細胞
肥大密切相關,并且證實這種促進心肌細胞肥大的效應是由TRPC介導的Ca2 +-NFAT信號通路完成[7]。在CaN被持續啟動后,小鼠心肌細胞膜TRPC6表達量明顯增加;在人類心力衰竭的心肌細胞中,TRPC6亦出現類似過表達。Nishida等[10]研究發現,內皮素-1( ET-1)、AngⅡ刺激體外大鼠心肌細胞和心肌成纖維細胞,可以顯著提高TRPC6的表達水平;血小板凝結抑制劑Forskolin可以顯著下調TRPC6表達; ET-1的這種作用是由Gα12/13( G-12家族蛋白的亞單位)所介導的,并且這種作用通過NFAT的持續激活持續啟動。進一步研究發現,過表達TRTRPC6的轉基因小鼠NFAT的轉錄活性增強,并參與Ca2 +-CaN/NFAT信號通路的正回饋機制,通過抑制5型磷酸二酯酶途徑抑制TRPC6表達活性增加造成的病理性心肌肥大[11]。高糖通過增加人類足突細胞黏連蛋白聚糖-4( SDC-4)和活性氧簇( ROS)的表達調節TRPC6的表達[12]。沒有基礎心臟病的Klotho基因缺陷小鼠會出現應激狀態下的心肌細胞肥大和心室重構;在健康成年小鼠的心肌細胞中,Klotho通過下調TRPC6而發揮心臟保護作用[13]。如果抑制Klotho基因缺陷小鼠TRPC6蛋白的表達,則可以阻止壓力誘導的心肌細胞重構。進一步研究發現,TRPC6高表達的小鼠會自發出現心肌重構;而Klotho過表達則可以明顯抑制TRPC6導致的心肌重構,延長小鼠生存期。Kinoshita等[14]證明,TPRC6蛋白是心鈉肽/腦鈉肽-鳥苷酸環化酶A ( ANP/BNP-GC-A)信號通路介導的心肌肥大作用的關鍵靶點。上述研究均證明,TRPC6在心肌肥厚的發生、發展過程中發揮重要的橋梁作用。
高糖可使心肌細胞內Na+-K+-ATP酶、Ca2 +-Mg2 +活性減低,使心肌細胞對鈣的攝取和輸送能力下降,導致心肌收縮功能受損。但高糖是否會通過上調TRPC蛋白的表達,導致胞內鈣超載,從而啟動CaN/NFAT信號通路,目前國內外尚無相關研究予以證實。
H9C2細胞源于胚胎期大鼠心臟,具有心肌細胞的很多特性; 33 mmol/L葡萄糖作為研究心肌細胞高糖損傷的條件,已經被廣泛證實并應用[15]。本研究將H9C2心肌細胞在33 mmol/L葡萄糖條件下培養,細胞出現肥大、變性、壞死,增殖率降低,說明高糖造成細胞損傷。在培養72 h時,3組TRPC6、NFAT3 mRNA及蛋白相對表達量較1、2組增高;加用TRPC通道阻滯劑skf96365后,心肌細胞增殖率有所增加,TRPC6、NFAT3 mRNA及蛋白相對表達量下調。上述結果提示,在高糖條件下TRPC6可能通過CaN/NFAT3通路造成心肌細胞損傷。
綜上所述,高糖抑制心肌細胞增殖,造成心肌細胞損傷;促進心肌細胞TRPC6表達,導致TRPC通道開放,引起心肌細胞內鈣超載,啟動CaN/NFAT3通路,可能是其作用機制。
參考文獻:
[1]Hamby RI,Zoneraich S,Shrman L.Diabetic cardiomyopathy[J].JAMA,1974,229( 13) : 1749-1754.
[2]Chavali V,Tyagi SC,Mishra PK.Predictors and prevention of diabetic cardiomyopathy[J].Diabetes Metab Syndr Obes,2013,( 6) : 151-160.
[3]席曉慧,王福文,牟艷玲.糖尿病心肌病的藥物治療研究進展[J].山東醫藥,2014,54( 5) : 93-95.
[4]簡春燕,吳鏗.糖尿病性心肌病發病機制和治療的研究現狀[J].中華臨床醫師雜志(電子版),2012,6( 1) : 164-167.
[5]Kumar S,Kain V,Sitasawad SL.High glucose-induced Ca2 +overload and ox-idative stress contribute to apoptosis of cardiac cells through mitochondrial de-pendent and independent pathways[J].Biochim Biophys Acta,2012,1820( 7) : 907-920.
[6]Dhalla NS,Takeda N,Rodriguez-Leyva D,et al.Mechanisms of subcellular remod-eling in heart failure due to diabetes[J].Heart Fail Rev,2014,19( 1) : 87-99.
[7]Bugger H,Abel ED.Molecular mechanisms of diabetic cardiomyopathy[J].Diabetologia,2014,57( 4) : 660-671.
[8]Eder P,Molkentin JD.TRPC channels as effectors of cardiac hypertrophy[J].Cire Res,2011,108( 2) : 265-272.
[9]Onohara N,Nishida M,Inoue R,et al.TRPC3 and TRPC6 are essential for angiotensinⅡ-induce cardiac hypertrophy[J].EMBO,2006,25( 22) : 5305-5316.
[10]Nishida M,Onohara N,Sato Y,et al.Gα12/13-mediated up-regulation of TRPC6 negatively regulates endothelin-1-induced cardiac myofibrob-last formation and collagen synthesis through nuclear factor of activated T cells activation[J].J Biol Chem,2007,282 ( 32) : 23117-23128.
[11]Nishida M,Watanabe K,Sato Y,et al.Phosphorylation of TRPC6 channels at Thr69 is requered for anti-hypertrophic effects of phosphodiesterase 5 inhibiton[J].J Biol Chem,2010,285 ( 17) : 13244-13253.
[12]Thilo F,Lee M,Xia S,et al.High glucose modifies transient receptor potential canonical type 6 channels via increased oxidative stress and syndecan-4 in human podocytes[J].Biochem Biophys Res Commun,2014,450( 1) : 312-317.
[13]Xie J,Cha SK,An SW,et al.Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart[J].Nat Commun,2012( 3) : 1238.
[14]Kinoshita H,Kuwahara K,Nishida M,et al.Inhibition of TRPC6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A signaling in the heart[J].Circ Res,2010,106( 12) : 1849-1860.
[15]Yu T,Sheu SS,Robotham JL,et al.Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species[J].Cardiovasc Res,2008,79( 2) :341-351.
收稿日期:( 2014-12-30)
通信作者:楊榮禮,E-mail: yrl6502@ sina.com
基金項目:徐州市科技計劃項目( KC14SH110)。
文章編號:1002-266X( 2015) 28-0024-03
文獻標志碼:A
中圖分類號:R541
doi:10.3969/j.issn.1002-266X.2015.28.008
