HPLC法同時測定氨咖黃敏膠囊中二組分的含量
2016-01-31 01:07:53程愛平
實用藥物與臨床 2015年12期
關鍵詞:高效液相色譜
鄒 燕,程愛平,林 燦
HPLC法同時測定氨咖黃敏膠囊中二組分的含量
鄒燕1*,程愛平1,林燦2
[摘要]目的建立高效液相色譜法同時測定氨咖黃敏膠囊中對乙酰氨基酚、咖啡因2種成分的含量。方法采用Agilent extend-C18(150 mm×4.6 mm,5 μm)色譜柱,流動相為乙腈(A)-三乙胺溶液(1 mL三乙胺加79 mL水,調pH至3.1,B)(17∶83),流速1.0 mL/min,檢測波長217 nm;柱溫25 ℃,進樣量15 μL。結果對乙酰氨基酚、咖啡因的線性濃度范圍分別為831.33~2 078.33 μg/mL (r=0.999 6)和50.27~125.67 μg/mL (r=0.999 4),兩組分平均回收率分別為99.99% (RSD=0.02%)和100.14% (RSD=0.52%)。結論該方法靈敏度、準確度高,重復性好,適用于氨咖黃敏膠囊中對乙酰氨基酚和咖啡因2種有效成分的測定。
[關鍵詞]氨咖黃敏膠囊;對乙酰氨基酚;咖啡因;高效液相色譜;含量測定
[Abstract]ObjectiveTo establish a method for simultaneous determination of paracetamol and caffeine in paracetamol,caffeine,artificial cow-bezoar and chlorphenamine maleate capsules by HPLC.MethodsThe HPLC system consisted of Agilent extend-C18column (150 mm×4.6 mm,5 μm) with a mobile phase of acetonitrile (A)-triethylamine solution (1 mL triethylamine+ 79 mL water,pH 3.1,B) (17∶83),the flow rate was 1.0 mL/min and the detection wavelength was at 217 nm.The column temperature was 25 ℃,and the sample quantity was 15 μL.ResultsThe linear ranges of paracetamol and caffeine were 831.33~2 078.33 μg/mL (r=0.999 6) and 50.27~125.67 μg/mL (r=0.999 4),and the average recovery rates were 99.99% (RSD=0.02%) and 100.14% (RSD=0.52%).ConclusionThe method is of high sensitivity,accuracy and repeatability,it can be used for the determination of paracetamol and caffeine in paracetamol,caffeine,artificial cow-bezoar and chlorphenamine maleate capsules.
收稿日期:2015-03-28
通信作者*
DOI:10.14053/j.cnki.ppcr.201512021
Content determination of two components in paracetamol,caffeine,artificial cow-bezoar and chlorphenamine maleate capsules by HPLCZOU Yan1*,CHENG Ai-ping1,LIN Can2(1.Jiaxing Institiute for Food and Drug Control,Jiaxing 314000,China;2.Jiaxing University,Jiaxing 314000,China)
Key words:Paracetamol,caffeine,artificial cow-bezoar and chlorphenamine maleate capsules;Paracetamol;Caffeine;HPLC;Content determination
0引言
氨咖黃敏膠囊是感冒用藥類非處方藥,適用于緩解普通感冒及流行性感冒所引起的發熱、頭痛、打噴嚏、咽喉疼痛、鼻塞等癥狀。本品為復方制劑,主要有效成分為對乙酰氨基酚、咖啡因、馬來酸氯苯那敏和人工牛黃。該藥物的質量標準收錄在《中國藥品標準》的化學藥品第三冊中[1]。在國家藥品標準中,對乙酰氨基酚的含量測定采用回流后外標指示劑法,咖啡因則采用氯仿萃取后滴定法測定,這兩種方法操作較繁瑣、存在的不確定因素較多。本文主要通過高效液相色譜法同時測定對乙酰氨基酚、咖啡因兩種成分的含量,結果表明方法可行,現報道如下。
1儀器與試藥
1.1儀器Agilent1200高效液相色譜儀(安捷倫公司);UV-2501PC紫外分光光度計(日本島津公司);XS105DU電子天平(梅特勒-托利多有限公司);Delta320H酸度計(梅特勒-托利多有限公司);超聲波清洗儀(USC502,上海波龍電子設備有限公司)。
1.2藥品與試劑氨咖黃敏膠囊(標示量為對乙酰氨基酚250 mg/粒、咖啡因15 mg/粒,浙江康恩貝股份有限公司,批號:20140313、20140827、20141001),對乙酰氨基酚對照品(含量100%,中國藥品生物制品檢定所,批號:100018-200408),咖啡因對照品(含量100%,中國藥品生物制品檢定所,批號:171215-201211)。磷酸(杭州化學試劑有限公司)分析純,乙腈(德國德姆施塔市墨客股份有限公司)色譜純,三乙胺[賽默飛世爾科技(中國)有限公司]色譜純。
2方法與結果
2.1分析方法驗證
2.1.1色譜條件色譜柱:Agilent extend-C18(150 mm×4.6 mm,5 μm);流動相:A相為乙腈,B相為三乙胺-水(1∶79),A∶B為17∶83;流速:1.0 mL/min;檢測波長:217 nm;柱溫:25 ℃;進樣量:15 μL。在該色譜條件下對照品中兩種成分的色譜峰分離度>1.5,理論塔板數均>2 000,符合使用條件。
2.1.2儲備溶液的配制精密稱取咖啡因對照品7.54 mg置10 mL容量瓶中,加流動相超聲5 min溶解后定量稀釋至刻度,搖勻,即得儲備液;放入4 ℃冰箱保存備用。精密稱取對乙酰氨基酚對照品12.47 mg置于10 mL容量瓶中,精密加入上述咖啡因對照品的儲備溶液1 mL,超聲3 min溶解后用流動相定容得混合對照品溶液。
2.1.3供試品溶液的配制取氨咖黃敏膠囊30粒,取內容物精密稱定,研細,精密稱取適量(約相當于對乙酰氨基酚250 mg),置于20 mL容量瓶中,加入少量流動相超聲溶解10 min,冷卻后定容。用濾紙過濾后,精密量取1 mL續濾液至10 mL容量瓶稀釋至刻度,即得供試品溶液。
2.1.4線性范圍取混合對照品溶液,依次精密吸取10、12、15、17、20、22、25 μL混合對照溶液注入高效液相色譜儀,記錄色譜圖。見圖1。以峰面積(Y)為縱坐標、對照品濃度(X,μg/mL)為橫坐標,分別繪制標準曲線,計算回歸方程。得對乙酰氨基酚方程Y=3.718 1 X+7 606.1 (r=0.999 6),線性范圍831.33~2 078.33 μg/mL;咖啡因方程Y=40.252 X+1 186.2 (r=0.999 4),線性范圍50.27~125.67 μg/mL。
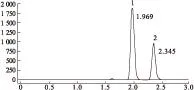
圖1 HPLC色譜圖
2.1.5精密度試驗取“2.1.2”項下混合對照品溶液,按“2.1.1”項條件,連續平行進樣5次。結果顯示,對乙酰氨基酚峰面積的RSD為0.21%,咖啡因峰面積的RSD為0.15%。
2.1.6穩定性試驗為了考察溶液的穩定性,取“2.1.3”項下供試品溶液,分別于0、2、4、24 h,按“2.1.1”項下色譜分析條件進行測定,記錄下2種成分的相應峰面積。結果對乙酰氨基酚RSD為0.56%,咖啡因RSD為1.22%,表明該樣品溶液在24 h內穩定性良好。
2.1.7重復性取“2.1.3”項下供試品溶液,按照 “2.1.1”條件連續進樣5次,記錄兩組分相應峰面積,得對乙酰氨基酚的RSD為0.3%,咖啡因為1.8%,表明兩組分的重復性良好。
2.1.8加樣回收率精密稱取對乙酰氨基酚對照品1 260.00 mg、咖啡因對照品75.04 mg置50 mL量瓶中,加流動相溶解并稀釋至刻度,搖勻;精密稱取已知含量的供試品適量(約相當于對乙酰氨基酚250 mg) 3份,分別置于20 mL容量瓶中,各加入上述對照品混合溶液8、10、12 mL,加入少量流動相超聲溶解10 min,冷卻后定容。用濾紙過濾后,精密量取1 mL續濾液置20 mL容量瓶加流動相稀釋至刻度,得加樣回收率溶液。按“2.1.1”項色譜條件進行分析,計算含量,結果見表1。
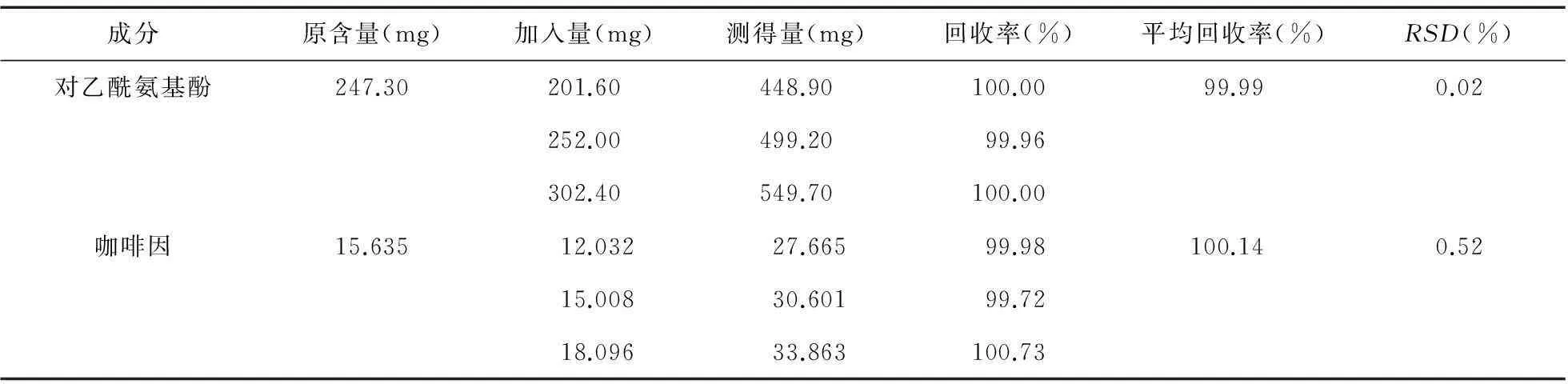
表1 回收率試驗結果
2.2樣品測定取康恩貝公司生產的3批不同的氨咖黃敏膠囊內容物按“2.1.3”項方法制成3份供試品溶液,注入高效液相色譜儀,按“2.1.1”項下色譜條件對3個供試品進行色譜分析,得到實驗數據,計算得到3批樣品中2種成分的百分含量。同時,按照國家藥品標準對含量進行測定,并與色譜法所得的結果進行比較,見表2。兩種方法實驗結果較為接近,相比較而言色譜法耗時少,操作簡便。
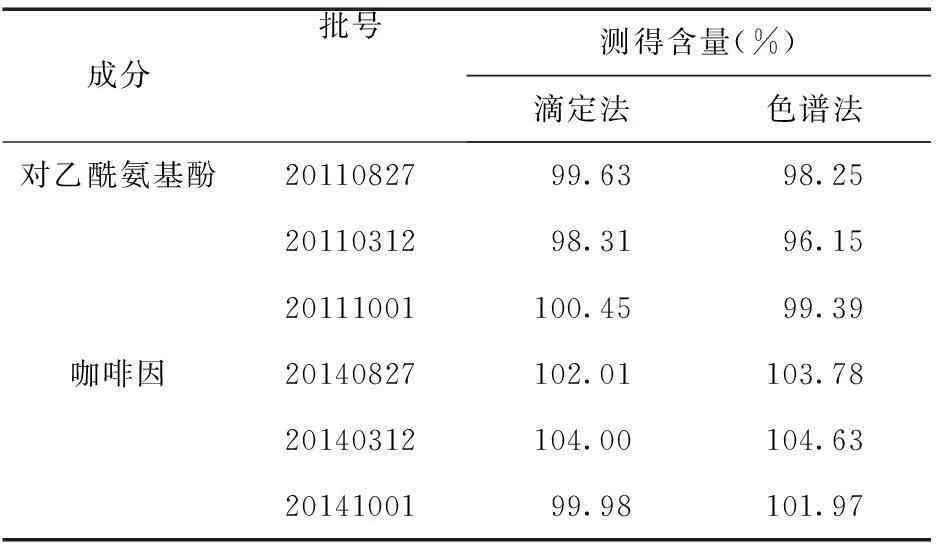
表2 樣品含量測定結果(n=3)
3討論
3.1波長的選擇根據相關文獻資料[2-5],選用常用的十八烷基鍵和硅膠色譜柱;另選定乙腈-三乙胺溶液(1 mL三乙胺加79 mL水,用磷酸調pH至3.1)(17∶83)為初始流動相,對2種成分的對照品進行溶解,然后用于紫外掃描。因每粒膠囊中含有對乙酰氨基酚250 mg、咖啡因15 mg,含量相差較大,為了避免在含量測定時對乙酰氨基酚濃度過高出現平峰或者咖啡因濃度過低不易出峰,選擇的波長應以Abs值咖啡因>對乙酰氨基酚為宜,且兩者的Abs差值盡量大。
取對乙酰氨基酚對照品、咖啡因對照品,用流動相制得含對乙酰氨基酚對照品8 μg/mL的溶液和含咖啡因對照品10 μg/mL的溶液。用 UV-2501PC紫外分光光度計在200~300 nm波長處對兩種溶液進行波長掃描,見圖2。由圖2可見,在約200~230 nm波長處,對乙酰氨基酚的Abs先于217.38處達到最小值,然后逐漸變大,且與咖啡因的Abs差值較大,故選擇217 nm作為測定波長。
3.2流動相比例的調整選擇乙腈為A相、三乙胺溶液(三乙胺∶水為1∶79,溶液用磷酸調pH至3.1抽濾)為B相,通過工作站調整有機相與水相的比例并保持其他條件不變。結果顯示,不同流動相比例下,各峰的分離度均>1.5,最終選定分離度>3且保留時間相對較短的流動相比例,即A∶B為17∶83。

圖2 UV圖譜
3.3柱溫的選擇筆者分別對日常實驗中最為常用的25、30、35 ℃進行了試驗,結果顯示,溫度對該樣品中所測2種成分含量沒有顯著影響,所以選擇藥品檢驗中較為常用的柱溫(25 ℃)作為該項實驗柱溫。
3.4流速的選擇為了選定適宜的流速,分別選用常用流速0.5、1、1.5 mL/min進行試驗,結果顯示,當流速為1.5 mL/min時,各峰的理論板數明顯降低,該流速舍去不考慮。流速為0.5、1 mL/min時,各參數均符合要求,此次選擇較為常用的1.0 mL/min。
本法選定的色譜條件效率高,分離度好,柱效理想;樣品的前處理較國家標準更簡單、快速;實驗結果與傳統方法所得實驗結果相近。因此,采用HPLC法同時測定氨咖黃敏膠囊中的對乙酰氨基酚和咖啡因的含量是可行的,為該藥品標準的修訂提供了參考。
參考文獻:
[1]國家藥典委員會.國家藥品標準化學藥品地方標準上升國家標準[S].2002:263.
[2]孫超,崔瀟,成秉辰,等.HPLC法同時測定氨咖黃敏膠囊中三種成分[J].哈爾濱商業大學學報(自然科學版),2013,29(2):145-147,175.
[3]吳俊芳,張濤,李麗萍.高效液相色譜法測定氨咖黃敏膠囊的含量[J].中國藥業,2011,20(4):33-34.
[4]張英芳,李明霞,韓春英.HPLC法同時測定氨咖黃敏膠囊中三組分的含量[J].齊魯藥事,2012,21(8):462-463.
[5]陳玉璞,張毅,董智攀.高效液相色譜法同時測定氨咖黃敏膠囊中對乙酰氨基酚和馬來酸氯苯那敏的含量[J].中國醫藥導報,2010,7(4):38-39.
作者單位:1.中國醫科大學藥學院,沈陽 110001;2.遼寧省建平縣醫院藥局,朝陽 122400;3.遼寧省中藥研究所,沈陽 110041
猜你喜歡
分析化學(2017年1期)2017-02-06 21:32:17
中國醫藥導報(2016年30期)2016-12-28 12:18:02
熱帶農業科學(2016年10期)2016-12-12 01:52:56
分析化學(2016年7期)2016-12-08 00:57:07
上海醫藥(2016年21期)2016-11-21 23:14:07
中國科技博覽(2016年18期)2016-10-19 11:09:28
科學與財富(2016年28期)2016-10-14 04:01:52
中國科技博覽(2016年2期)2016-04-25 14:06:58
上海醫藥(2016年3期)2016-03-23 23:38:20
現代儀器與醫療(2015年4期)2015-07-15 10:13:19
