吡唑醚菌酯的合成研究
2016-02-04 12:49:40劉東志周雪琴
化學工業與工程 2016年1期
李 清,李 巍,劉東志,周雪琴
(天津大學化工學院,天津300072)
甲氧基丙烯酸酯類殺菌劑是一類低毒、高效、廣譜、內吸性殺菌劑,是世界農藥界極具發展潛力和市場活力的新型農用殺菌劑[1]。吡唑醚菌酯(Pyraclostrobin)又名唑菌胺酯[2],是目前活性最高的甲氧基丙烯酸酯類殺菌劑,由德國巴斯夫公司于1993年開發研究,并于2002年在歐洲市場推出,與氟環唑復配用于防治谷物病害,在50多個國家登記100多種作物,也可用于非農作物[3]。該藥具有高效、低毒、對環境友好、適用作物廣泛等特點。自吡唑醚菌酯上市以來的短短幾年,該品種的市場迅速飆升,銷售額迅速上升,已列為所有菌劑品種市場的第 2位,僅次于嘧菌酯[4]。
吡唑醚菌酯在美國、歐洲等國家擁有專利權,在我國也申請了專利,其合成技術尚未公開,研究其合成工藝對改善我國的落后的農藥結構,降低傳統殺菌劑對環境的污染有著非常重要意義。綜合文獻其合成方法主要歸為以下兩種,均以鄰硝基甲苯和對氯苯胺為起始原料,反應主要包括環合、氧化、還原、酰化、甲基化、溴化和縮合共七步生成最終目標物[5-11]。合成路線主要有兩種,如圖1所示。
中間體1-(4-氯苯基)-3-吡唑醇的合成路線如圖2所示[12-13]。
本研究采用了合成路線二,對鄰硝基甲苯先進行還原、酰化、甲基化后進行溴化,由于強吸電子基團硝基消失,故路線二較路線一溴化更容易一些。本研究對整個工藝進行優化,并對關鍵步驟溴化進行了改進,提出溴化反應結束后未經提取產物,直接進入下一步縮合反應,原因是一溴代物與剩余原料及副產物二溴代物分離相當困難,而且通過研究表明對于下一步縮合和反應整體收率影響不大,且減少了操作程序,提高了效率。該工藝具有反應原料易得,條件溫和,產品純度高等優點,適合工業化生產。
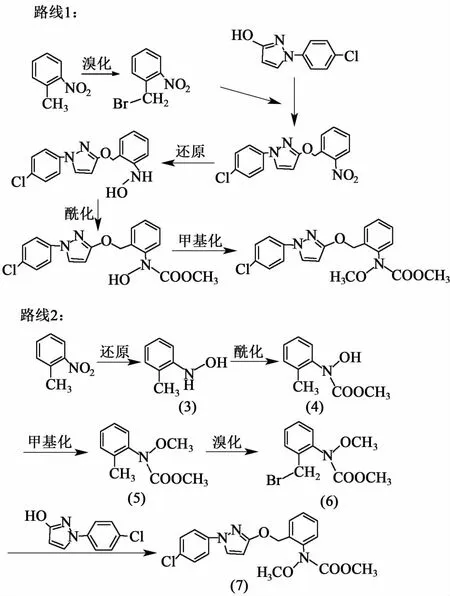
圖1 合成路線示意圖Fig.1 Schematic scheme of synthesis
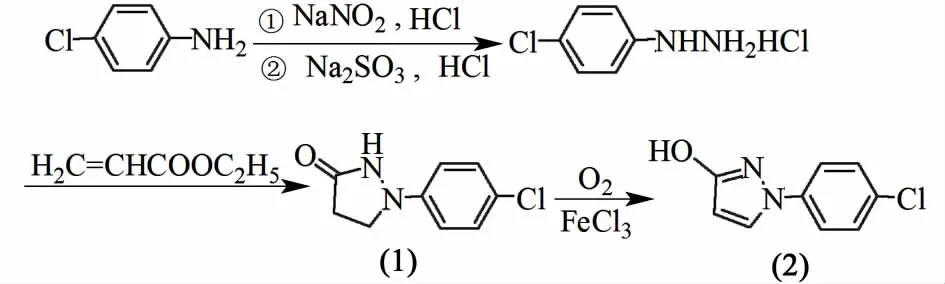
圖2 中間體合成路線示意圖Fig.2 Schematic scheme of the synthesis of intermediate
1 試驗部分
1.1 試劑與儀器
對氯苯胺和氯甲酸甲酯為工業品,其他試劑均為市售分析純。
1H-NMR由美國Burker AV500型核磁共振儀測定(TMS內標,CDCl3或CD3OD為溶劑);質譜由英國VG公司 VGZAB-HS型質譜儀測定;熔點由YRT-3型藥物熔點儀測定。
1.2 合成
1.2.1 1-(4-氯苯基)-3-吡唑醇的合成
1.2.1.1 對氯苯肼鹽酸鹽的合成
250mL三口瓶中依次加入對氯苯胺38.4 g(0.301 mol)、工業濃鹽酸 110 g(0.903 mol)、水144 g,5℃以下滴加亞硝酸鈉溶液[亞硝酸鈉22.8 g(0.330 mol) +水 98 g],滴畢反應 30 min,過濾得重氮鹽溶液。
500mL三口瓶中依次加入亞硫酸鈉94.5 g(0.75 mol)、水 400 g,10 ℃左右滴加重氮鹽溶液,滴完反應30 min后,加鋅粉 2 g(0.0357 mol),升溫至70℃反應3 h,熱過濾,將濾液升至80℃,滴加工業濃鹽酸進行酸化,滴畢80℃下反應2 h,冷卻至室溫,有白粉色固體析出,過濾得產品 55.0 g,純度97.5%(LC),收率 99.5%,熔點 223.5~228.5℃(分解),文獻值 225 ~230 ℃[14]。
對氯苯肼鹽酸鹽用氫氧化鈉溶液中和制得對氯苯肼,核磁測試結果如下:1H-NMR(500 MHz,CD3Cl)δ:6.75(d,2H,-H-Ar),7.17(d,2H,-H-Ar),5.2(s,1H,-NH),3.57(s,2H,-NH2)。對氯苯肼鹽酸鹽 ESI(m/z):142.0(M+)。
1.2.1.2 1-(4-氯苯基)吡唑烷-3-酮(1)的合成
250mL三口瓶中加入無水乙醇200 g,稱取固體鈉10.5 g(0.456 mol),現制乙醇鈉溶液,40℃下加入對氯苯肼鹽酸鹽 27.5 g(0.153 mol),攪拌30 min后,滴加丙烯酸甲酯 17.5 g(0.203 mol),30 min滴畢,保溫反應1.5 h后升溫回流反應4.0 h,減壓去除溶劑得粗品,加適量水溶解固體,10%乙酸溶液調pH值至5~6,冷卻過濾得產品19.0 g,純度97.0%(LC),收率 61.0%。
1H-NMR(500 MHz,CD3OD)δ:8.5(s,1H,-NH),7.0(d,2H,-H-Ar),7.3(d,2H,-HAr),3.9(t,2H,-CH2),2.5(t,2H,-CH2)。
1.2.1.3 1-(4-氯苯基)-3-吡唑醇(2)的合成
500mL三口瓶中依次加入吡唑酮19.65 g(0.1 mol),三氯化鐵 2.36 g(0.0145 mol),質量分數為5%的氫氧化鉀溶液200 g,通空氣,1.0 h升溫至80℃,反應8.0 h氧化完全,加大量水,冷卻至室溫,用鹽酸酸化至 pH值為1~2,有淺黃色固體析出,冷卻過濾得產品19.7 g,純度97.7%(LC),收率99.0%。
1H-NMR(500 MHz,CD3OD)δ:10.3(w,1H,-OH),7.6(d,2H,-H-Ar),7.4(d,2H,-HAr),7.9(d,1H,-CH),5.8(d,1H,-CH)。
1.2.2 N-羥基-N-2-甲基苯基氨基甲酸甲酯(4)的合成
500mL三口瓶中依次加入鄰硝基甲苯31.2 g(0.228 mol),二氯乙烷250 g,氮氣保護下加入鎳粉5.5 g(0.0932 mol),0 ~5 ℃下滴加水合肼 32 g(0.467 mol),30 min滴畢,氮氣保護攪拌反應 6~7 h,過濾去除鎳粉,鹽酸調pH值至4~5,萃取分層取有機相,加100 g水,5~10℃下滴加氯甲酸甲酯21 g(0.222 mol),30 min 滴畢,保溫反應 1.0 h,萃取分層取有機相,減壓蒸干溶劑,50℃下加甲苯80 g,攪拌30 min,冷卻析出固體,抽濾得白色固體36.5 g,純度 99.0%(LC),收率為87.6%。
1H-NMR(500 MHz,CD3Cl)δ:8.26(w,1H,-OH),7.2 ~7.4(m,4H,-H-Ar),3.77(s,3H,-CH3),2.33(s,3H,-CH3)。
1.2.3 N-甲氧基-N-2-甲基苯基氨基甲酸甲酯(5)的合成
1 000mL三口瓶中依次加入 N-羥基-N-2-甲基苯基氨基甲酸甲酯31 g(0.171 mol),無水碳酸鉀35 g(0.254 mol),二氯甲烷 600 g,攪拌加熱至 58 ~60 ℃,滴加硫酸二甲酯 23.7 g(0.188 mol),30 min滴畢,保溫反應6.0 h后,加適量水,升溫至70℃,保溫1.0 h后停止加熱,用水洗滌萃取3次,取有機層減壓蒸溶劑,得橙黃色液體33.5 g,純度98.2%(LC),收率 99.3%。
1H-NMR(500 MHz,CD3Cl)δ:7.2 ~7.4(m,4H,-H-Ar),3.74 (s,3H,-CH3),3.69(s,3H,-CH3),2.25(s,3H,-CH3)。
1.2.4 N-甲氧基-N-2-溴甲苯氨基甲酸甲酯(6)的合成
500mL三口瓶中依次加入N-甲氧基-N-2-甲苯氨基甲酸甲酯 19.5 g(0.1 mol),過氧化苯甲酰(BPO)1.21 g(0.05 mol),NBS18.15 g(0.102 mol),四氯化碳180mL,回流反應5 h,抽濾,蒸干溶劑得橙黃色黏稠液體33.8 g,液相測目標產物含量為66.9%。
1.2.5 吡唑醚菌酯(7)的合成
500mL三口瓶中依次加入1-(4-氯苯基)-吡唑醇15.56 g(0.080 mol),無水碳酸鉀 20.7 g(0.15 mol),丙酮 150mL,四丁基溴化銨 0.82 g(0.0030 mol),升溫至回流,將上一步所得N-甲氧基-N-2-溴甲苯氨基甲酸甲酯溶于50mL丙酮滴加至三口瓶中,3.0 h滴畢,回流反應6.0 h,抽濾,減壓蒸干溶劑,加入無水甲醇90mL,攪拌至全溶,冷卻有晶體析出,過濾得淺黃色固體17.8 g,純度98.4%(LC),收率56.5%,熔程 61.5~62.9 ℃(文獻值63.7~65.2 ℃[3])。
1H-NMR(500 MHz,CD3Cl)δ:7.36 ~7.67(m,8H,-H-Ar),7.69(d,1H,-CH),5.93(d,1H,-CH),5.35(s,2H,-CH2),3.80(s,3H,-CH3),3.77(s,3H,-CH3);ESI(m/z):315(M+1)。
2 結果與討論
2.1 對氯苯肼鹽酸鹽的合成
2.1.1 還原反應
還原反應中加入鋅粉可以使還原完全,同時使副產物中偶氮化合物的氮氮雙鍵還原為單鍵,使偶氮化合物顏色變淺,提高外觀質量。本研究在其它條件不變情況下,考察了鋅粉的加入量對反應的影響,結果見表1。

表1 鋅粉對產物的影響Table 1 Influence of zinc on product
實驗表明,鋅粉的加入量為對氯苯胺質量的0.05時,已能明顯提高外觀質量,少于0.03達不到外觀要求,大于0.05無進一步改觀。
2.1.2 酸化反應
用鹽酸進行酸析時,有明顯的放熱現象,同時反應放出大量的SO2氣體,可使產物氧化,本研究考察了酸化溫度對于產物的影響,結果見表2。

表2 還原溫度對收率的影響Table 2 Influence of reduction temperature on yield
結果表明,酸析溫度高于85℃使副產物明顯增加,若溫度低于80℃,酸析不完全,酸析溫度為80~85℃時,產物收率最佳。
2.2 1-(4-氯苯基)-3-吡唑酮的合成
1)溶劑的選擇:在反應時間為6 h,其他條件不變的情況下,分別考察了甲苯、乙醇、甲苯和乙醇混合體作溶劑對反應的影響,結果見表3。

表3 溶劑對產物的影響Table 3 Influence of solvent on product
結果表明乙醇作溶劑,所得產品質量含量及收率最高。由于甲苯沸點較乙醇沸點高,在乙醇回流溫度下反應副產物少,反應速率快,回流反應4 h幾乎完畢,而且乙醇相對較環保。對于甲苯和乙醇混合體系,所得產品質量含量和收率相對較低,而且單一溶劑有利于回收利用。
2)反應溫度對收率的影響:吡唑酮是由苯肼和酯經過麥克加成、合環兩步反應合成,對于合環反應的溫度做了對比實驗研究,結果見表4。

表4 反應溫度的影響Table 4 Influence of reaction time on yield
結果表明:反應在78~80℃下反應,產物收率較高。溫度太低,苯肼和酯不能充分環合,反應不徹底。溫度太高,反應物和溶劑容易分解且產物收率下降。
綜上所述,較優反應條件為:乙醇作溶劑,在回流溫度下反應4 h,產品含量97.0%(LC),收率61.0%。
2.3 1-(4-氯苯基)-3-吡唑醇合成的影響因素討論
1)溶劑的選擇:此步反應屬于氧化反應,對于反應溶劑本研究選用了不同質量分數的KOH水溶液,DMF和NaOH水溶液做了對比研究,結果見表5。

表5 溶劑對產物的影響Table 5 Influence of solvent on product
結果表明,當溶劑選用質量分數為5%的KOH水溶液時,產品純度和收率較高,選用質量分數為10%和15%的KOH作溶劑時,產品質量含量和收率沒有明顯的提高。對于DMF作溶劑,產品含量及收率相對低,從環保的角度來考慮,水溶液更環保一些;這些溶劑反應所需時間基本相同,約7~8 h,故本實驗選用質量分數為5%的KOH水溶液作溶劑。
2)催化劑三氯化鐵用量的影響結果見表6。

表6 FeC l3用量對產物的影響Table 6 Influence of FeC l3 amount on product
結果表明,m(FeCl3)/m(吡唑酮)為0.08 時,產品含量及收率都相對較低,0.16時,產物含量及收率無近一步提升,0.12時產物含量及收率較高。
綜上所述,較優反應條件為:5%KOH的水溶液作溶劑,m(FeCl3)/m(吡唑酮)為0.12,產品含量97.7%(LC),收率 99.0%。
2.4 N-羥基-N-2-甲基苯基氨基甲酸甲酯合成的影響因素
此步反應必須在氮氣保護下進行,否則鎳粉很容易被氧化從而失去活性。反應溫度應在0~5℃,大于5℃硝基會被直接還原為氨基,使產品質量含量和收率低。催化劑鎳粉的加入量對反應影響也很大,本研究考察了鎳粉的加入量對反應時間的影響,結果見表7。

表7 鎳粉加入量對反應的影響Table 7 Influence of nickel amount on reaction
催化劑的量偏低會導致反應速率較慢,實驗結果表明n(鎳粉)/n(鄰硝基甲苯)為0.48時,反應時間相對較短,大于0.48,時間沒有明顯縮短,小于0.48,反應較慢,時間較長。
綜上所述,較優反應條件為0~5℃下,氮氣保護,n(鎳粉)/n(鄰硝基甲苯)為0.48,產品含量99.0%(LC),收率 87.6%。
2.5 N-甲氧基-N-2-甲基苯基氨基甲酸甲酯的合成
常用的甲基化試劑有硫酸二甲酯和碳酸二甲酯,本研究考察了這兩種甲基化試劑加入量對反應的影響,結果見表8。
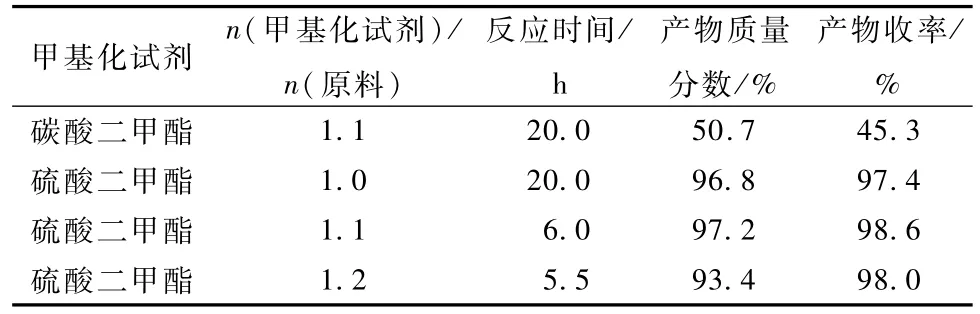
表8 甲基化試劑對反應的影響Table 8 Influence of methylating reagent on reaction
實驗結果表明,選用碳酸二甲酯因其活性低,故副反應多,產品質量含量和收率低,且反應時間較長。選用硫酸二甲酯,反應速度快,幾乎沒有副反應發生。n(硫酸二甲酯)/n(原料)為1.1時產品質量含量及收率高,產品顏色淺,反應時間短;為1.2時,反應時間沒有太多的縮短,但因為其活性高而發生副反應,導致產品顏色較深;為1.0時,反應較慢,所用時間較長。
綜上所述,硫酸二甲酯作甲基化試劑,n(硫酸二甲酯)/n(原料)為1.1,6 h反應完畢,所得產品含量98.2%(LC),收率99.5%。
2.6 N-甲氧基-N-2-溴甲苯氨基甲酸甲酯的合成
此步反應屬于自由基反應,對于自由基反應的影響因素很多,本研究從引發劑的種類及其用量,溴化劑這幾個影響因素進行了研究。
1)在 n(N-甲氧基-N-2-甲苯氨基甲酸甲酯)∶n(NBS)∶n(引發劑) =1.00∶1.02∶0.50,四氯化碳作溶劑,回流反應5 h的條件下考察了引發劑AIBN和BPO對反應的作用效果,結果見表9。

表9 引發劑對反應的影響Table 9 Influence of initiator on reaction
因為AIBN分解溫度是64℃,其使用溫度范圍一般在45~65℃,80℃左右會急劇分解,其1 h半衰期溫度是82℃。而BPO一般在60~80℃分解,故在四氯化碳回流溫度下引發劑BPO作用效果遠比AIBN好。
2)在 n(N-甲氧基-N-2-甲苯氨基甲酸甲酯)∶n(NBS) =1.00∶1.02,四氯化碳作溶劑,回流反應5 h的條件下考察了BPO的用量對反應的影響,結果見表10。
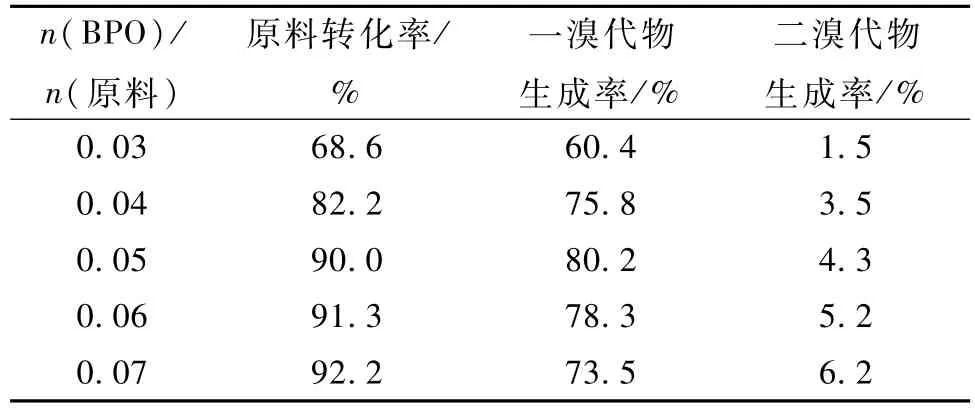
表10 BPO對反應的影響Table 10 Influence of BPO on reaction
結果表明,n(BPO)/n(原料)為0.05時,原料轉化率及目標產物收率相對較高;大于0.05時,導致二溴代物的生成率提高;小于0.05時,原料轉化率及一溴代物生成率都較低。因為自由基反應中,引發劑量不足會使自由基生成速率低,從而使反應速率慢,原料轉化率低。過量則自由基生成速率快,反應速率也快,導致副產物二溴代物生成率提高,另外過量的引發劑也會導致自由基猝滅而影響反應。
3)溴化劑的選擇:溴化劑常用的有溴素,溴化氫及NBS,本研究選用NBS,其特點是可以持續的提供低濃度的液溴。雖然可以用稀釋的液溴代替,但液溴存取不方便,且稀釋的程度不易控制,故選用NBS較適宜。n(NBS)/n(原料)為1.02,過量會導致二溴代物生成率增加,不足原料轉化率低。由于未反應的原料、一溴代產物和二溴代產物不好分離,故將得到的粗品直接進入下一步反應,結果表明對最終產物的收率影響不大,且可以減少操作。
通過實驗研究,較適宜物料配比為n(N-甲氧基-N-2-甲苯氨基甲酸甲酯)∶n(NBS)∶n(過氧化苯甲酰)=1.00∶1.02∶0.50,四氯化碳作溶劑,回流(79~80℃)反應5 h,過濾,蒸干溶劑得橙黃色黏稠液體33.8 g,液相測目標產物66.9%。
2.7 吡唑醚菌酯的合成
對于產品的結晶溶劑,本研究選用異丙醇,乙醇,甲醇做了對比研究,結果見表11。

表11 結晶溶劑對產物的影響Table 11 Influence of crystallization solvent on product
結果表明,甲醇效果較佳,而且價格比異丙醇低,降低成本。
通過實驗研究,最適宜物料配比為n(N-甲氧基-N-2-溴甲苯氨基甲酸甲酯)∶n(1-(4-氯苯基)-3-吡唑醇)∶n(無水碳酸鉀)∶n(四丁基溴化銨)=1.00∶1.00∶1.50∶0.03,丙酮做溶劑,回流反應 6 h,用甲醇后處理后所得產品純度為98.4%(LC),收率58.5%。
3 結論
對吡唑醚菌酯的合成工藝進行了研究改進:以對氯苯胺為原料經過重氮化、還原、酸化及氧化四步反應合成中間體 1-(4-苯基)-3-吡唑醇,LC測得含量為97.7%,總收率為60.7%;以鄰硝基甲苯為原料,經過還原,酰化,溴化后再與中間體1-(4-苯基)-3-吡唑醇反應生成目標化合物吡唑醚菌酯,LC測得純度大于98.4%,總收率大于56.7%(以鄰硝基甲苯計),比現有文獻有了較大提高[5,12],并對合成的關鍵步驟溴化做了優化改進,減少了副產物的生成,提高收率,降低成本,整個反應過程操作簡單,較適合工業化生產,具有很好的應用前景。
[1]張國生.甲氧基丙烯酸酯類殺菌劑的應用、開發現狀及展望[J].農藥科學與管理,2003,(12):30-34 Zhang Guosheng.Application,present development situation and prospects of the Strobilurins fungicides[J].Pesticide Science and Administration,2003,(12):30-34 (in Chinese)
[2]柏亞羅.Strobilurins類殺菌劑研究開發進展[J].農藥,2007,46(5):290-305 Bo Yaluo.Research and development progress of Strobilurins fungicides[J].Pestices,2007,46(5):290-305(in Chinese)
[3]Ammermann E,Lorenz G,Scgekberger K.In the british crop protection council conference pest&disease[M].British Crop Protection Council,Brighton UK,2000,541-548
[4]趙平,嚴秋旭,李新,等.甲氧基丙烯酸酯類殺菌劑的開發及抗性發展現狀[J].農藥,2011,(8):547-551,572 Zhao Ping,Yan Qiuxu,Li Xin,et al.Development and current situation of the resistance development of Strobilurins fungicides[J].Pestices,2011,(8):547-551,572 (in Chinese)
[5]陶賢鑒,羅亮明,黃超群,等.吡唑醚菌酯的合成研究[J].農藥研究與應用,2009,(1):16-17,21 Tao Xianjian,Luo Liangming,Huang Chaoqun,et al.Synthesis of pyraclostrobin.[J].Research and Application of Pesticides,2009,(1):16-17,21 (in Chinese)
[6]Call H P.Process for deligification of a lignin containing pulp:US,6103509[P].2000-08-15
[7]Lorenz G,Muelller B.Canbamates and crop protection agents containing them:WO,9315046[P].1993-08-05
[8]李仲英,李江勝,劉衛東.N-甲氧基-N-2-甲基苯基氨基甲酸甲酯的合成工藝研究[J].湖南大學學報,2004,2(1):4-6 Li Zhongying,Li Jiangsheng,Liu Weidong.Synthesis of N-methoxy-N-2-methyl phenyl carbamate[J].Journal of Hunan University,2004,2(1):4-6(in Chinese)
[9]Freudenreich J,Stohrer J.Method for producing organic hydroxylam ines:US,6211410[P].2001-04-03
[10]Chapman DW,Caskey D C.Process for the preparation of arylhydroxylamines:EP,0086363 [P].1983-08-24
[11]張永臣.唑菌胺酯的合成工藝研究[D].哈爾濱:黑龍江大學,2009 Zhang Yongchen.Synthesis of pyraclostrobin[D].Haerbin:Hei Longjiang University,2009 (in Chinese)
[12]Henry H B.Improvement in the manufacture of hydroxyammo compounds:GB,493960[P].1938-10-18
[13]Davis G C.Moderated reduction reactions for producing arylhydroxvlamine:US,4723030[P].1988-02-02
[14]潘富友,楊健國,梁華定,等.對氯苯肼鹽酸鹽的合成工藝[J].應用化學,2001,(12):1001-1003 Pan Fuyou,Yang Jianguo,Liang Heading,et al.The synthesis of p-chlorophenylhydrazine hydrochloride[J].Applied chemistry,2001,(12):1001-1003 (in Chinese)
