一類申報新藥雷騰舒工作對照品核磁共振定量分析方法的建立
2016-02-22 10:30:59賈國慧呼海濤
上海醫藥 2016年1期
賈國慧++呼海濤


摘要 目的:建立一類申報新藥雷騰舒工作對照品的核磁共振定量方法。方法:以咖啡因為內標,采用核磁共振定量法對雷騰舒樣品進行定量。結果:本方法三個不同濃度水平的樣品平均回收率為99.87%,RSD0.76%。重復性考察RSD為0.53%。日間精密度考察RSD為0.20%。結論:所建立的核磁共振定量方法,簡便、快速、準確性、重復性好,可作為該新藥工作對照品定量的方法之一。
關鍵詞 核磁共振定量法 雷騰舒 工作對照品賦值
中圖分類號:TQ460.72; 0657.2
文獻標識碼:A
文章編號:1006-1533(2016)01-0072-03
隨著我國新藥研究的發展,企業研發人員需要為開發的新藥制備工作對照品并進行定量的情況越來越多。為確保定量的可靠性,往往需要用多種不同原理來建立定量方法。通過不同方法所測數據的差異,對工作對照品定量的準確性與可靠性進行確認。常用的方法豐要包括色譜扣除法(質量平衡法)和容量分析方法等,然而對于有些沒什么滴定活性基團的化合物,往往很難找到合適的容量分析方法。
本研究為擬申報化學11類新藥雷騰舒(研發代號T8,目前已在II期臨床評價階段,圖1)藥學研究中的一部分,按照新藥研究指導原則,須采用不同原理的方法對其工作對照品進行定量(賦值)研究。在我們完成質量平衡法評估后,希望采用經典的容量分析方法進行比對研究。然而通過對本品結構、性質的深入分析,發現其結構中存在的3類可能的可滴定基團(環氧基、羥基、內酯環)都很難設計量化方案。分析如下。
①環氧基結構中存在的三個環氧結構單元,各自的化學環境不盡相同,難以量化反應程度。②羥基結構中有兩個羥基,一個為二級羥基、另一個為三級羥基。二級羥基雖可通過與醋酐反應酰化后,用堿滴定牛成的醋酸,從而完成定量,但前提是須確保酰化完全。即使可以酰化完全,醋酐也很容易使三級羥基脫水,從而干擾定量結果。③內酯環該基團理論上可與堿反應,先加成再脫去-OR,但因本品內酯環上同時存在雙鍵,會導致1、2加成與l、4加成同時存在,很難量化。預試驗結果也證實了上述分析。
面對上述問題,如何選擇更加便捷、可靠的賦值方法成為我們在新藥申報中必須考慮的問題。通過調研,我們發現采用已知含量的其他化合物做內標的定量核磁方法可以很好地解決我們所面臨的問題。方法具有專屬性好、無需已知含量自身對照品的技術特點。
為了建立并驗證該方法在新藥一雷滕舒樣品定量上的可行性,我們對該方法進行了研究,并獲得了準確性、重復性良好的結果。
1 材料與方法
1.1 儀器
核磁共振儀(德國布魯克,Ascend 400)。天平(瑞士梅特勒,XP56,百萬分之一)。
1.2 試藥
雷騰舒(本公司自制,批號:235-61、20080102);咖啡因(Alfa公司,含量99.6%);氘代乙腈、氘代吡啶(J&K Scientific Ltd.);氘代二甲亞砜(Cambridge Isotope Laboratories. Inc.)。
1.3 樣品制備與計算
精密稱定咖啡因(約4mg)及本品(約6mg)適量至2ml量瓶,以氘代乙腈溶解并定容至刻度,搖勻,取約0.5ml到核磁管,上樣測定。分別記錄化學位移值為3.345、3.358組峰(T8)的面積和化學位移值為3.274峰(咖啡因)的峰面積,按下述公式計算。
A1/A2=n1W1/M1/(n2W2/M2)
其中A1為T8定量峰的面積、A2為內標咖啡因定量峰的面積;
n1為T8定量峰氫質子數(n1=1),n2為咖啡因定量峰氫質子數(n2=3);
W1為T8測定量,W2為咖啡因稱樣量,M1、M2分別為T8、咖啡因的分子量。
1.4 核磁共振儀條件設置
核磁共振儀:2930序列,共振頻率400兆赫,磁場強度9.4T,弛豫時間12s,中心頻率3.31ppm。
2 結果
2.1 方法的重復性
采用本法對經干燥的T8樣品(批號:235-61)平行測定5次,考察方法的重復性(表1)。結果表明,RSD小于2,方法重復性很好。
2.2 方法的重現性
為了考察方法的重現性,我們在不同日期,對上述T8樣品進行了測定,經計算方法重現性好(表2)。
2.3 方法的準確度
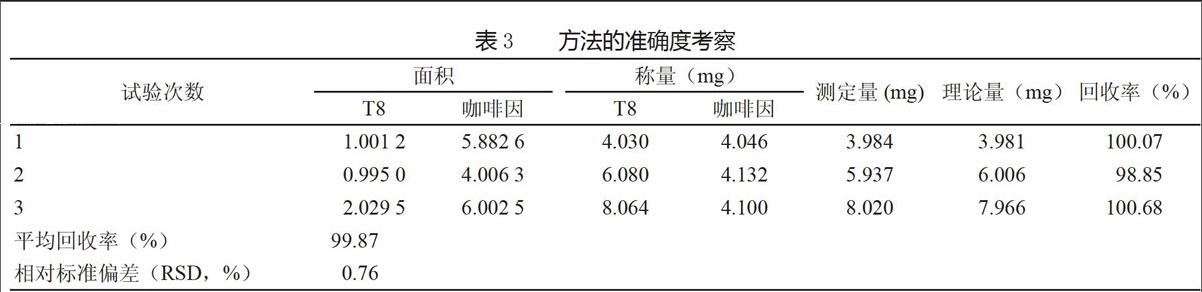
采用上述方法,對方法準確度進行考察。取經干燥的T8原料(批號:235-61)4、6、8mg,精密稱定,分別置于2ml量瓶中,再分別加入精密稱定的4mg咖啡因,用氘代乙腈溶解并稀釋至刻度,搖勻,吸取0.5ml至核磁管中,測定。從平均回收率和RSD值可見該方法準確度非常好(表3)。
2.4 與質量平衡法數據的比較
采用本法和質量平衡法分別檢測了批號為20080102的T8樣品。本法測定結果:99.20%、質量平衡法測定結果:99.56%。兩種方法測定結果基本一致。
3 討論
工作對照品定量(賦值)是研發工作中經常遇到的問題。在追求真值的過程中,需要多種分析于段協同作用,綜合判斷。采用核磁共振儀進行定量,提供了不同原理定量的義一方法。該方法的定量研究,國內外已有報道,其工作原理也在多國藥典上有詳述,但在新藥附值研究的實際應用方面報道不多。
我們在方法建立過程中發現,內標選擇、樣品前處理、儀器參數設置等因素,都會對檢測結果產牛影響,須關注。
以本研究為例,我們篩選了多個化合物,最終選定了咖啡因(圖1)作為內標物。因為咖啡因結構中氫的磁場環境比較簡單,可找到與本研究被測物峰位不干擾且場位比較接近的測量峰;另外,經考察,咖啡因在我們選定的溶解樣品的溶劑氘代乙腈中非常穩定而且不會與被測物反應。這些特點保障了測定結果的穩定性和重復性。
新藥賦值研究中,節約樣品和選擇合適的溶解樣品的溶劑也是我們方法設計中不得不考慮的問題。為篩選到樣品溶解度高、穩定性好、且對測定無干擾的溶劑,我們分別考察了氘代二甲亞砜、氘代吡啶、氘代乙腈三種溶劑,結果發現,氘代二甲亞砜為溶劑,雖然可滿足溶解度高、溶劑干擾少、樣品穩定性好等要求,但重復性、線性結果不穩定,無法達到預期的定量要求。經分析發現,為節省樣品與氘代試劑,我們在試驗方案中選用了1ml量瓶進行定容,由于氘代二甲亞砜黏度大,容易導致定量誤差,使測定結果重現性不好。為此,我們換用氘代吡啶作為溶解溶劑對方法進行考察,發現T8在氘代吡啶中不穩定,放置后溶液顏色會發牛變化,測定結果可靠性差。通過這兩次溶解溶劑對測定結果的影響,我們重新分析了T8的溶解度、穩定性,最終選擇氘代乙腈為溶劑,獲得了比較理想的結果。
此外,儀器的參數設置,也是影響數據穩定性的重要因素,特別是中心頻率、弛豫時間、掃描次數的設定。合適的中心頻率(參數OI)的選擇,可使脈沖對各定量峰強度的影響差異最小。另外,須考察弛豫時間、掃描次數對外加標準和待測物響應穩定性的影響,以確定最佳參數。
綜上,在新藥開發過程中無法獲得標準品對照的情況下,采用核磁定量法對企業自制工作對照品進行純度分析與賦值,是不同原理進行純度評估的新選擇。其優點除簡單、快捷之外,還具有不需要已知含量待測樣品的對照品,僅需要已知含量的普通化學品的特點。這很好地解決了新藥工作對照品賦值研究中無對照品的問題,且測定數據可靠性好。
