R59022調節心肌細胞自噬促進ET-1誘導的乳鼠心肌細胞肥大
2016-03-24 01:28:37柳玉梅張貴平張海寧
中國藥理學通報 2016年2期
關鍵詞:自噬
柳玉梅,尹 園,張貴平,張海寧
(廣州醫科大學藥理教研室,廣東 廣州 511436)
?
R59022調節心肌細胞自噬促進ET-1誘導的乳鼠心肌細胞肥大
柳玉梅,尹園,張貴平,張海寧
(廣州醫科大學藥理教研室,廣東 廣州511436)
中國圖書分類號:R-332;R322.11;R329.24;R542.202.2;R977.3
摘要:目的研究甘油二酯激酶(DGK)的抑制劑R59022對心肌細胞自噬及內皮素 1 (ET-1) 誘導的心肌肥大的影響,并探討其可能機制。方法原代培養乳鼠心肌細胞,ET-1誘導乳鼠心肌細胞肥大;Western blot法檢測自噬相關蛋白微管相關蛋白1輕鏈3 (LC3)、beclin-1、p62的表達以及Akt的磷酸化及非磷酸化蛋白表達;RT-PCR技術檢測心肌肥大基因腦鈉肽(BNP)、β-肌球蛋白重鏈(β-MHC)的mRNA表達水平;細胞免疫熒光檢測心肌細胞表面積。結果ET-1作用乳鼠心肌細胞24 h明顯促進心肌細胞肥大,并誘導自噬相關蛋白LC3-II/I、beclin-1的表達增強,p62表達減弱。自噬抑制劑氯喹(CQ)、3-甲基腺嘌呤(3-MA)減小心肌細胞表面積,下調肥大基因BNP、β-MHC的mRNA表達,改善ET-1誘導的心肌細胞肥大,而自噬激動劑雷帕霉素(RAPA)則促進ET-1誘導心肌細胞肥大;R59022預處理增加ET-1誘導的LC3-II/I、beclin-1的表達,增強ET-1誘導的心肌細胞自噬,同時進一步促進ET-1誘導的心肌細胞表面積的增大,BNP、β-MHC的mRNA表達水平的增加,促進ET-1誘導的心肌細胞肥大;對Akt表達的結果顯示,ET-1明顯下調Akt的磷酸化水平,R59022對此有促進作用。結論R59022增強ET-1誘導的心肌細胞自噬,促進心肌細胞肥大,其機制可能與抑制Akt的激活,從而抑制mTOR通路的活化有關。
關鍵詞:內皮素1;心肌細胞;心肌肥大;自噬;甘油二酯激酶;R59022
心肌肥大(cardiac hypertrophy)是心臟應對長期壓力超負荷緩慢產生的一種代償機制,常伴隨蛋白質合成增加、心臟結構重構和細胞器的功能障礙,主要表現為心肌細胞體積增大、膠原纖維形成的肌節增加及胚胎基因的再表達,是先天性心臟病、高血壓、心肌梗死、心力衰竭等多種心血管疾病共有的病理過程。自噬(autophagy)是指細胞在自噬相關基因(autophagy related gene,Atg)的調控下,利用溶酶體降解自身受損的蛋白質和細胞器的過程。自噬是生物維持蛋白代謝平衡及細胞環境穩定的共同機制,廣泛參與了各種生理和病理過程[1]。研究表明[2-3],自噬參與調控心肌肥大,但其具體作用及機制目前尚不明確。
G蛋白偶聯受體(G protein coupled receptors,GPCRs)/甘油二酯(diacylglycerol,DAG)/蛋白激酶C(protein kinase C,PKC)信號通路在調控心肌肥大的發展進程中發揮非常重要的作用。甘油二脂激酶(diacylglycerol kinase,DGK)是DAG的磷酸激酶,通過磷酸化DAG,減少DAG的可利用性,可以削弱DAG對PKC的激活,是負調節磷脂酰肌醇信號通路的關鍵信號分子。我們以往的研究顯示[4],DGK在心肌組織分布的主要亞型DGK-ζ 明顯抑制內皮素1(endothelin-1,ET-1)誘導的心肌肥大,但其機制尚未完全闡明。新近有研究表明,DGK的抑制劑R59022能誘導神經細胞株NG108-15的自噬[5],提示DGK可能通過調節心肌細胞自噬,進而影響心肌肥大。為研究DGK 調節的心肌細胞自噬在心肌肥大中的作用,本實驗首先觀察了 R59022對ET-1誘導的原代乳鼠心肌細胞肥大的影響,并探討其可能分子機制。
1材料與方法
1.1材料
1.1.1動物出生1~3 d的清潔級SD大鼠,♀♂不拘,廣東省醫學實驗動物中心提供,許可證:SCXK粵2013-0002。
1.1.2主要試劑高糖DMEM、胎牛血清(FBS)購自Gibco;ET-1、R59022、5-溴脫氧尿嘧啶核苷(BrdU)、氯喹(chloroquine,CQ)、三甲基腺嘌呤(3-methyl adenine,3-MA)、胰蛋白酶、F-actin-鬼筆環肽等試劑均購自Sigma;雷帕霉素(rapamycin, RAPA)購自上海浩然生物技術有限公司;LC3、beclin-1、p62、p-Akt、Akt 、p-AMPK、AMPK等抗體以及ECL發光液均購自Cell Signaling Technology;β-actin購自Bioworld;逆轉錄相關試劑購自TaKaRa;PCR引物由Invitrogen公司合成。
1.2方法
1.2.1新生大鼠原代心肌細胞分離、培養及藥物處理出生1~3 d SD大鼠,無菌開胸取心臟,剪碎,胰酶反復多次消化后,收集消化的心肌細胞懸液,于4 ℃離心收集細胞。用含質量分數為10% FBS的DMEM重懸細胞,差速貼壁法除去貼壁的成纖維細胞,未貼壁的心肌細胞以合適密度接種于細胞培養皿或孔板中,置于細胞培養箱內培養24 h,換無血清培養基饑餓24 h后,以ET-1(10-7mol·L-1)作用細胞24 h誘導乳鼠心肌細胞肥大,CQ(5 μmol·L-1)、3-MA(5 mmol·L-1)、RAPA(10 nmol·L-1)和R59022(10-5mol·L-1)分別于加入ET-1前0.5 h加入,以觀察其對自噬相關蛋白表達及心肌肥大的影響。
1.2.2Western blot法檢測蛋白表達藥物處理后,用RIPA裂解液裂解細胞,提取總蛋白。BCA法測定蛋白濃度后,取等量蛋白行SDS-PAGE凝膠電泳,電泳結束后將蛋白轉移至PVDF膜上,剪取目的條帶,以脫脂奶粉室溫封閉1 h,隨后加入LC3、beclin-1、p62、p-Akt、Akt、β-actin一抗4 ℃孵育過夜,TBST洗膜后,室溫孵育相應二抗1 h,TBST洗膜后,用ECL試劑盒檢測蛋白表達水平,Quantity one軟件分析灰度值,以目的條帶的灰度值與β-actin的灰度值的比值表示目的蛋白的表達水平。
1.2.3細胞免疫熒光檢測心肌細胞表面積藥物處理后,除去培養基,細胞經固定、破膜、封閉后,用TRITC-鬼筆環肽避光孵育以標記F-actin,于熒光顯微鏡下觀察并記錄結果。每組隨機選取30個細胞,用圖像分析軟件Image-Pro Plus 6.0計算心肌細胞表面積。
1.2.4RT-PCR法檢測心肌肥大標記物BNP、β-MHC的mRNA表達水平TRIzol裂解細胞提取總RNA,逆轉錄成cDNA后,取逆轉錄產物進行PCR擴增,條件:95 ℃變性 3 min,95 ℃ 30 s,55 ℃ 30 s,72 ℃ 30 s,共35個循環, 72 ℃ 延伸 10 min。反應完成后,取PCR產物行瓊脂糖凝膠電泳(含溴化乙錠0.5 mg·L-1),用BI2000凝膠成像系統分析,以目的基因條帶的積分光密度值(IOD)與β-actin的 IOD之比確定目的基因BNP、β-MHC的mRNA表達水平。
2結果
2.1ET-1誘導心肌細胞自噬如Fig 1所示,給予ET-1分別作用心肌細胞0~12 h,ET-1對自噬相關蛋白LC3-II/I、beclin-1及p62表達沒有明顯影響,但與0 h相比,ET-1作用細胞24 h明顯誘導LC3-II/I、beclin-1表達上調(P<0.05,Fig1A、1B),并使p62表達明顯降低(P<0.05,Fig 1B);預先給予自噬溶酶體的抑制劑CQ抑制溶酶體降解后,較未經CQ預處理細胞相比,ET-1作用各時間點LC3-II/I的表達有增加趨勢,且在24 h明顯增加(P<0.01,Fig 1A),表明ET-1明顯誘導心肌細胞自噬及自噬流。
2.2自噬抑制劑及激動劑對ET-1誘導的心肌細胞肥大的影響如Fig 2所示,與control組相比,ET-1明顯促進心肌細胞表面積增加(P<0.01,Fig 2A)及肥大基因BNP、β-MHC的mRNA表達水平的上調(P<0.01,Fig 2B、2C),誘導心肌細胞肥大;自噬抑制劑CQ、3-MA對上述指標發揮明顯抑制作用(P<0.01,Fig 2A、2B),而自噬激動劑RAPA則發揮明顯的促進作用(P<0.01,Fig 2A、2C)。提示自噬激活促進ET-1誘導的心肌細胞肥大。
2.3R59022促進ET-1誘導的心肌細胞自噬如Fig 3所示,與control組相比,ET-1作用于心肌細胞24 h,LC3-II/I及beclin-1表達明顯上調(P<0.05,P<0.01);R59022預處理明顯促進ET-1誘導的上述指標上調(P<0.05,P<0.01)。
2.4R59022促進ET-1誘導的心肌細胞肥大如Fig 4所示,R59022預處理促進ET-1誘導的心肌細胞表面積增加(P<0.01,Fig 4A)及BNP、β-MHC 的mRNA表達水平的上調(P<0.05,Fig 4B),促進ET-1誘導的心肌細胞肥大。
2.5R59022下調Akt的磷酸化水平如Fig 5所示,與control組相比,ET-1明顯下調Akt的磷酸化水平(P<0.05,Fig 5A),R59022預處理促進ET-1誘導的Akt磷酸化水平的下調(P<0.05,Fig 5A);而對AMPK的磷酸化水平,ET-1及R59022均未見明顯影響(P>0.05,Fig 5B)。
3討論
心肌肥大是心肌重構的關鍵組成部分,是導致心源性猝死和心力衰竭風險明顯升高的危險因素[6],其發生機制復雜,目前尚不完全明確。ET-1是公認的促肥大因子[7],通過與其特異性的受體結合,在介導心肌肥大的發生、發展過程中發揮重要作用[8]。本研究結果證實,ET-1作用原代乳鼠心肌細胞24 h明顯增大心肌細胞表面積,增加肥大基因BNP、β-MHC 的mRNA表達水平,誘導原代乳鼠心肌細胞肥大。
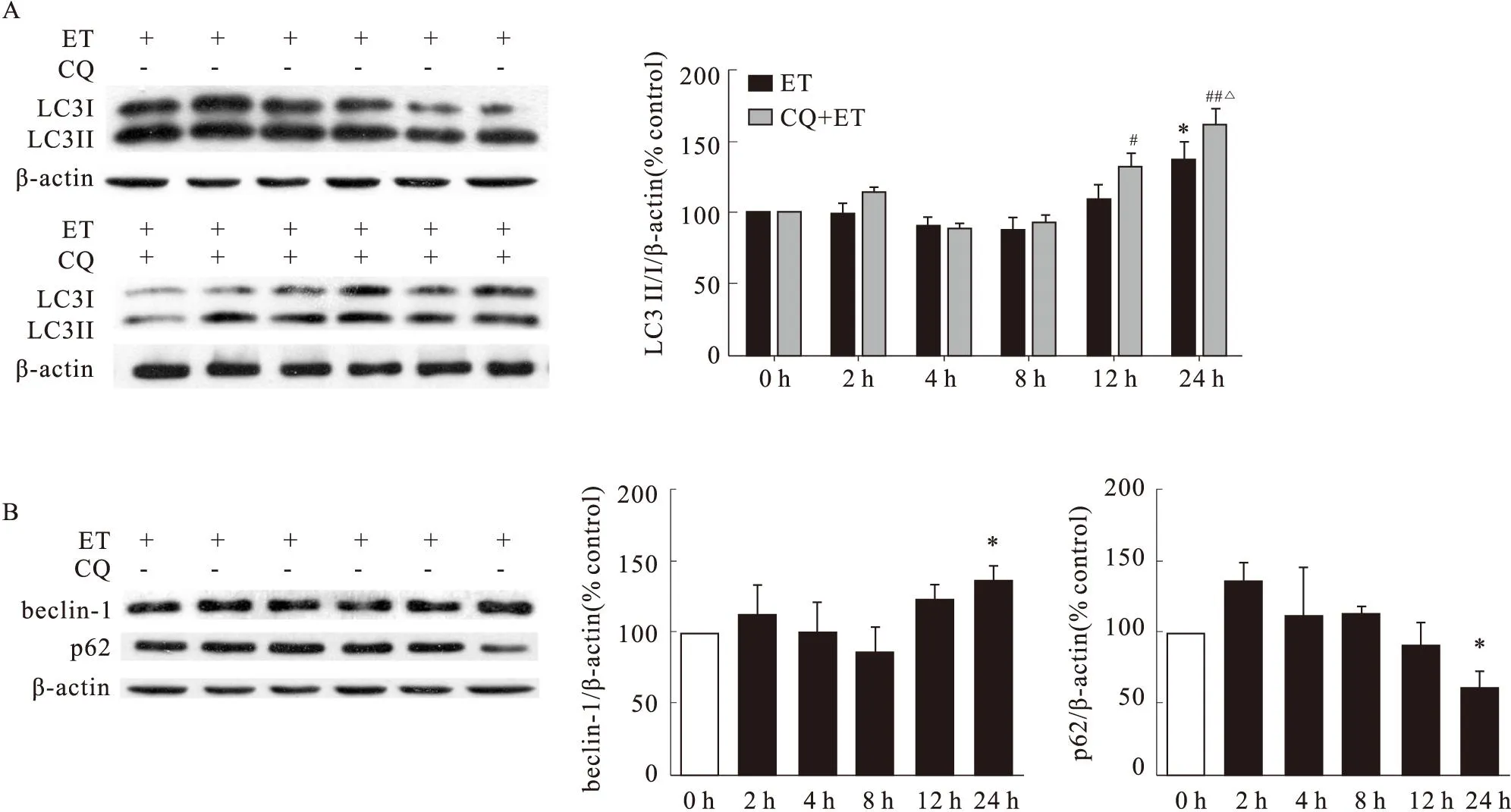
Fig 1 Effects of ET-1 on expression of autophagy-related proteins(n=3~8)
A: Effects of ET-1 on expression of LC3II/I in the presence or absence of CQ pretreatment; B:Effects of ET-1 on expression of beclin-1 and p62.*P<0.05vsET 0 h;#P<0.05,##P<0.01vsET+CQ 0 h;△P<0.05vsET 24 h

Fig 2 Effects of 3-MA,CQ and RAPA on ET-1-induced myocardial hypertrophy(n=3~4)
A: Effects of 3-MA,CQ,RAPA on cardiomyocyte size; B: Effects of 3-MA,CQ on mRNA levels of BNP and β-MHC; C: Effects of RAPA on mRNA levels of BNP and β-MHC.*P<0.05,**P<0.01vscontrol;##P<0.01vsET

Fig 3 Effects of R59022 on expression of autophagy-related proteins(n=6)

Fig 4 Effects of R59022 on ET-1-induced myocardial hypertrophy(n=3~4)
A: Effects of R59022 on cardiomyocyte size; B: Effects of R59022 on mRNA levels of BNP and β-MHC.*P<0.05,**P<0.01vscontrol;#P<0.05,##P<0.01vsET.

Fig 5 Effects of R59022 on expression of Akt and AMPK(n=4~5)
A:Effects of R59022 on expression of Akt; B: Effects of R59022 on expression of AMPK.*P<0.05vscontrol;#P<0.05vsET.
近年的研究顯示,自噬參與調控心肌肥大的發生發展,但其對心肌肥大的具體作用目前尚不明確。有研究認為自噬激活起促進心肌肥大的作用[9],也有研究認為自噬激活對心肌肥大起保護作用[10]。造成自噬在心肌肥大中作用不同,甚至相反的原因可能是疾病的誘因、損傷程度及自噬的水平不同。自噬的發生主要包括三個階段:第一階段,自噬誘導因素刺激內質網、高爾基體或細胞膜等形成自噬前體;第二階段,在Atgs的參與下,自噬前體延伸, 包裹需要降解的蛋白質和受損的細胞器, 形成自噬體;第三階段,自噬體與溶酶體結合形成自噬溶酶體,降解產物并參與機體代謝。從自噬前體的形成到內容物降解的整個流程即自噬流(autophagy flux)。在自噬的發展過程中,beclin-1是自噬啟動階段的關鍵因子,參與調控自噬前體的產生。在自噬被誘導后,由原位于胞質中的LC3I與磷脂酰乙醇胺(phosphatidyl ethanolamine,PE)偶合,形成始終穩定錨定在自噬體膜上的LC3II,標志著自噬體的形成,因此,研究者以往多以beclin-1、LC3蛋白的表達或者以綠色熒光蛋白標記LC3等方法間接反映自噬的活性。然而,自噬體數量的增加不能完全反映自噬的活性,因為造成自噬體數量增加的原因可能是自噬的激活,還可能是自噬溶酶體的降解障礙導致自噬體無法降解而堆積,所以新近研究多以自噬流評價自噬[11-12]。本研究中,我們檢測了自噬標志性蛋白LC3、beclin-1的表達,還檢測了可以反映自噬流通暢性的自噬相關蛋白p62,以及通過觀察CQ在溶酶體水平阻斷自噬的降解后LC3的表達水平來評價自噬水平。結果顯示,ET-1明顯誘導LC3II/I、beclin-1蛋白表達的增加,促進p62蛋白的降解。并且,在用CQ阻斷自噬降解后,LC3II/I的表達水平較未經CQ處理的細胞相比明顯增加,提示ET-1誘導心肌肥大過程中明顯伴隨自噬水平的上調。為了進一步明確自噬在ET-1誘導的心肌細胞肥大過程中的作用,本研究觀察了自噬抑制劑3-MA、CQ和自噬激動劑RAPA對ET-1誘導的心肌細胞肥大的影響。結果顯示,3-MA、CQ抑制自噬可以明顯改善ET-1誘導的心肌細胞肥大,而RAPA激活自噬則進一步促進ET-1誘導的心肌細胞肥大,提示自噬激活參與調節ET-1誘導的乳鼠心肌細胞肥大。
研究表明,Gq介導的磷脂酰肌醇信號通路在心肌肥大的發生、發展中發揮重要作用。DGK是磷脂酰肌醇信號通路的負調節因子,通過磷酸化DAG,可以削弱DAG對該信號通路的激活,在抑制心肌肥大發展過程中可能發揮關鍵作用,但其具體的胞內信號目前尚不完全清楚。有研究報道,DGK的抑制劑R59022能誘導神經細胞株NG108-15自噬[5]。我們對心肌纖維化的研究結果亦顯示[13],DGK參與調節心肌成纖維細胞自噬和纖維化,提示DGK亦可能通過參與調節心肌細胞自噬,進而影響心肌肥大過程。因此,本研究首先觀察了DGK的抑制劑R59022對心肌細胞肥大及自噬的影響。我們的結果顯示,R59022預處理促進ET-1誘導的心肌細胞肥大,同時對ET-1誘導的自噬相關蛋白LC3II/I、beclin-1的表達水平的上調亦有促進作用,提示R59022可能通過促進心肌細胞自噬,從而促進了ET-1誘導的心肌細胞肥大。
目前已經明確的調控自噬的信號通路主要分為哺乳動物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)依賴型途徑和非mTOR依賴型途徑。mTOR作為自噬啟動階段的關鍵調節因子,其活化后可抑制自噬的發生,是自噬的主要負調控因子[14]。目前認為mTOR信號通路上游的刺激因子主要是胰島素、生長因子、營養因子、能量以及壓力。生長因子和胰島素的刺激可以激活PI3K,PI3K進一步活化其下游關鍵效應因子Akt,Akt直接磷酸化mTOR的Ser2448 位點,激活mTOR,從而抑制自噬。磷酸腺苷激活蛋白激酶(AMP-activated kinase,AMPK)則是細胞能量感受的總開關,通過mTOR信號通路和非mTOR信號通路如p53、ULK1[15]、avicin D[16]等調控細胞自噬與凋亡,在細胞的代謝調控中發揮重要作用。本研究結果顯示,ET-1誘導乳鼠心肌細胞肥大過程中,Akt的磷酸化水平明顯下調,R59022對此有促進作用,但是ET-1和R59022并不影響AMPK的磷酸化水平,提示R59022增強ET-1誘導的心肌細胞自噬促進心肌細胞肥大的機制可能與抑制Akt的激活,從而抑制mTOR通路的活化有關,而AMPK信號可能并不參與R59022的自噬促進作用,但其分子機制尚有待進一步明確。
參考文獻
[1]林超,劉兆國,錢星,等. 自噬在心血管疾病中的作用研究進展[J].中國藥理學通報, 2014,30(10):1347-9.
[1]Lin C, Liu Z G, Qian X, et al. Research progress on the role of autophagy in cardiovascular diseases[J].ChinPharmacolBull, 2014,30(10):1347-9.
[2]Oyabu J,Yamaguchi O,Hikoso S,et al.Autophagy-mediated degradation is necessary for regression of cardiac hypertrophy during ventricular unloading[J].BiochemBiophysResCommun,2013,441(4):787-92.
[3]Dai D F,Rabinovitch P.Mitochondrial oxidative stress mediates induction of autophagy and hypertrophy in angiotensin-II treated mouse hearts[J].Autophagy,2011,7(8):917-8.
[4]Huang Y,Zhang H,Shao Z,et al.Suppression of endothelin-1-induced cardiacmyocyte hypertrophy by PPAR agonists:role of diacylglycerol kinase zeta[J].CardiovascRes,2011,90(2):267-75.
[5]Takita T,Konuma T,Hanazato M,Inoue H.Diacylglycerol kinase inhibitor R59022-induced autophagy and apoptosis in the neuronal cell line NG108-15[J].ArchBiochemBiophys,2011, 509(2):197-201.
[6]Frey N,Olson E N.Cardiac hypertrophy:the good,the bad,and the ugly[J].AnnuRevPhysiol,2003,65:45-79.
[7]Li H,Gao S,Ye J,et al.COX-2 is involved in ET-1-induced hypertrophy of neonatal rat cardiomyocytes: role of NFATc3[J].MolCellEndocrinol,2014,382(2):998-1006.
[8]孔令雷,顏玲娣,宮澤輝.內皮素系統與心肌肥厚[J].中國藥理學通報,2008,24(9):1127-30.
[8]Kong L L,Yan L D,Gong Z H.Endothelins system and myocardial hypertrophy[J].ChinPharmacolBull,2008,24(9):1127-30.
[9]Rawat D K,Alzoubi A,Gupte R,et al.Increased reactive oxygen species,metabolic maladaptation,and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure[J].Hypertension,2014,64(6):1266-74.
[10]Liu C,Xue R,Wu D,et al. REDD1 attenuates cardiac hypertrophy via enhancing autophagy[J].BiochemBiophysResCommun,2014,454(1):215-20.
[11]Lavandero S, Troncoso R,Rothermel B A,et al.Cardiovascular autophagy:concepts,controversies,and perspectives[J].Autophagy,2013,9(10):1455-66.
[12]Zhang Y,Xu X,Ren J.MTOR overactivation and interrupted autophagy flux in obese hearts:a dicey assembly[J]?Autophagy,2013,9(6):939-41.
[13]鄒剛玲,柳玉梅,張海寧.自噬對AngII誘導的心肌成纖維細胞增殖及遷移的影響及機制[J].廣東藥學院學報,2015,31(3):407-11.
[13]Zou G L,Liu Y M,Zhang H N.Effect of autophagy on AngⅡ-induced proliferation and migration of cardiac fibroblasts[J].JGuangdongPharmUniv,2015,31(3):407-11.
[14]Mizushima N,Levine B,Cuervo A M,Klionsky D J. Autophagy fights disease through cellular self-digest[J].Nature,2008,451(7182):1069-75.
[15]Wang S,Song P,Zou M H. AMP-activated protein kinase,stress responses and cardiovascular diseases[J].ClinSci,2012,122(12):555-73.
[16]Xu Z X,Liang J,Haridas V,et al.A plant triterpenoid,avicin D,induces autophagy by activation of AMP-activated protein kinase[J].CellDeathDiffer,2007,14(11):1948-57.
R59022 promotes ET-1-induced cardiac hypertrophy in neonatal rat cardiomyocytes via regulating autophagy
LIU Yu-mei,YIN Yuan,ZHANG Gui-ping,ZHANG Hai-ning
(DeptofPharmacology,GuangzhouMedicalUniversity,Guangzhou511436,China)
Abstract:AimTo investigate the effects of DGK inhibitor R59022 on ET-1-induced myocardial hypertrophy and autophagy, and explore the possible mechanisms. MethodsMyocardial hypertrophy was induced by ET-1 in cultured rat neonatal cardiomyocytes. Western blot was used to detect the expression of microtubule-associate protein 1 light chain 3(LC3), beclin-1, p62, p-Akt and Akt. mRNA expression of brain natriuretic peptide (BNP) and beta mysion heavy chain (β-MHC) and the cell size of cardiomyocytes were detected by RT-PCR and immunofluorescence, respectively. ResultsTreatment cardiomyocytes with ET-1(10-7mol·L-1) for 24 h induced the myocardial hypertrophy in cultured neonatal rat cardiomyocytes with the activation of autophagy as evidenced by the increased expression of autophagy-related proteins LC3-II/I and beclin-1, as well as the increased p62 degradation. While, myocardial hypertrophy induced by ET-1, including the increased myocardial cell size and the mRNA expression of fetal gene BNP and β-MHC, could be reversed by autophagy inhibitor 3-methyl adenine (3-MA) and chloroquine (CQ),but promoted by autophagy agonist rapamycin (RAPA).Pretreatment cardiomyocytes with R59022, an inhibitor of DGK, enhanced ET-1-induced myocardial hypertrophy by enhancing autophagy in cardiomyocytes.Furthermore,ET-1 treatment inhibited the activation of Akt by the down-regulation of the Akt phosphorylation, and R59022 enhanced the effect of ET-1 on the activation of Akt. ConclusionsEnhanced autophagy contributes to cardiomyocyte hypertrophy. R59022 deteriorate ET-1-induced myocardial hypertrophy by activating autophagy. The possible mechanism may be related to the inhibition of activation of mTOR signaling pathway by inhibiting the activation of Akt.
Key words:endothelin-1; cardiomyocyte; cardiac hypertrophy; autophagy; diacylglycerol kinase; R59022
文獻標志碼:A
文章編號:1001-1978(2016)02-0239-06
doi:10.3969/j.issn.1001-1978.2016.02.018
作者簡介:柳玉梅(1989-),女,碩士生,研究方向:心血管藥理學,E-mail:liuyumei1220@126.com;張海寧(1968-),女,博士,副教授,研究方向:心血管藥理學,通訊作者,E-mail:zhanghn_gz@126.com
基金項目:廣州市教育局基金資助項目(No 10A179)
收稿日期:2015-11-14,修回日期:2015-12-20
網絡出版時間:http://www.cnki.net/kcms/detail/34.1086.R.20160125.1557.036.html網絡出版地址:2016-1-25 15:57
猜你喜歡
中國醫藥導報(2017年2期)2017-03-18 20:50:25
中國中藥雜志(2017年1期)2017-03-06 21:37:10
中國中藥雜志(2016年21期)2017-02-16 13:30:44
中國中藥雜志(2016年21期)2017-02-16 12:24:39
糖尿病新世界(2016年16期)2016-12-09 04:07:18
中西醫結合心血管病電子雜志(2016年6期)2016-11-14 12:40:03
科技視界(2016年11期)2016-05-23 08:10:09
湖北農業科學(2015年20期)2015-11-12 21:01:15
中國醫藥導報(2015年15期)2015-08-07 01:10:32
山東體育學院學報(2015年2期)2015-05-27 13:17:09
