香榧轉錄組測序及生物信息學基礎分析
2016-03-31 07:07:54易官美包燕春1寧波城市職業技術學院浙江寧波31550江西省南昌市青山湖風景區管理處江西南昌330039
山東農業大學學報(自然科學版) 2016年1期
易官美,包燕春1.寧波城市職業技術學院,浙江寧波31550.江西省南昌市青山湖風景區管理處,江西南昌330039
?
香榧轉錄組測序及生物信息學基礎分析
易官美1*,包燕春2
1.寧波城市職業技術學院,浙江寧波315502
2.江西省南昌市青山湖風景區管理處,江西南昌330039
摘要:香榧具有重要的經濟價值,但其基因組信息相對匱乏,限制了其分子生物學和基因功能的研究。本文以不同組織的香榧作為研究對象,采用新一代高通量測序技術平臺Illumina HiSeq?2000對香榧轉錄組進行測序和數據分析,共得到37,349,086個reads片段,總堿基數為4.35 G。利用組裝軟件,對獲得的高質量序列進行組裝,共得到104,636個Unigene,平均長度為784 nt,N50為1,702。將Unigene序列與公共數據庫進行比對,28,766個Unigenes獲得了注釋。其中26,856個Unigene在NR蛋白數據庫中獲得注釋,24,003個Unigenes在NT數據庫中獲得注釋,21,401個Unigene在Swiss-Prot蛋白數據庫中獲得注釋,16,137個Unigene在COG數據庫中獲得注釋,11,410個Unigene在GO數據庫中獲得注釋。根據KEGG注釋信息,18,564個Unigene被劃分到256個代謝途徑中。SSR位點搜索發現,在4,217個Unigene中含有4,706個SSR位點。分析所獲得的轉錄組數據,將為香榧功能基因的克隆,基因的表達,指紋圖譜構建和分子標記輔助選育奠定基礎。
關鍵詞:香榧;轉錄組;高通量測序;SSR
香榧(Torreya grandis Fort. ex Lindl. cv. merrillii)為紅豆杉科(Taxaceae)榧樹屬(Torreya)榧樹(Torreya grandi Fort. ex Lindl.)的栽培變種。榧樹起源于中國,在中國的湖南、江西、安徽、江蘇、浙江和福建等省均有自然分布。榧樹是世界上比較稀有的經濟樹種之一,既可以食用,也可以藥用,還是綠化觀賞的常用樹種。香榧干果風味獨特,富含營養,有止咳化痰、潤肺明目、殺蟲殺菌等多種功效;另外,香榧中含有的紫杉醇是一種具有抗癌活性的生理活性物質[1,2]。
利用基因組、轉錄組及蛋白質組學等各種組學技術可以揭示細胞生理活動的規律、探明生物代謝的機理[3]。轉錄組是某一物種或特定的細胞在某一個時間段或特定的環境條件下產生的所有轉錄物的集合。利用轉錄組學技術可以了解到基因的功能和基因的結構,可以揭示器官以及細胞在特定生物學過程中的分子機制[4]。近年來,轉錄組學的研究應用廣泛,已應用于多種樹木,主要有紅豆杉(Taxus mairei)[5]、木欖(Bruguiera gymnorrhiza)[6]、剛毛檉柳(Tamarix hispida)[7]、桉樹(Eucalyptus robusta)[8]、日本落葉松(Larix leptolepis)[9]、油桐(Vernicia fordii)[10]、橡膠樹(Hevea brasiliensis)[11]和茶樹(Camellia sinensis)[12]等。
目前,對香榧的研究主要集中在生理生化特性、栽培技術[1,2]和居群遺傳學[13]等方面,缺少對其基因組學以及轉錄組學的研究。本研究利用Illumina高通量測序技術對香榧的轉錄組進行測序,利用生物學軟件將獲得的數據進行拼接組裝,再利用生物信息學技術將獲得的Unigene進行基因功能注釋、基因功能分類以及代謝途徑的分析,以期為香榧重要性狀基因的克隆及基因功能的分析、分子標記的開發以及遺傳圖譜的構建奠定良好的基礎。
1 材料和方法
1.1材料來源
香榧的葉片、幼果和枝干組織采集于浙江省寧波市奉化市錦溪村香榧種植基地(北緯29°32′24",東經121°11′50.8",海拔540 m)屬亞熱帶季風性氣候,四季分明,溫和濕潤,年均氣溫16.3℃,最高氣溫40.5℃,最低氣溫為-11.1℃。年平均降水量1500 mm。日照時數1850 h,無霜期232 d,生長于海拔500 m左右的山坡。采集8年生正常健壯的香榧嫁接苗,同一株香榧個體上的葉片,幼果,枝干組織各2 g,進行等量混樣。立即用液氮速凍,在-80°C冰箱中冷凍保存備用。
1.2方法
1.2.1RNA提取總RNA的提取方法按照Invitrogen公司的Trizol Reagent說明書進行。DNA的消化處理按Promega公司DNaseⅠ的方法進行。RNA完整性檢測利用Agilent的BioAnalyzer 2 100,樣品RIN≥8。
1.2.2轉錄組測序及序列組裝香榧cDNA文庫的構建采用TruSeq RNA Sample Preparation Kit (Illumina)進行。利用帶有Oligo(dT)的磁珠對mRNA進行富集。然后將打斷成短片段的mRNA為模板,合成cDNA的第一條鏈。然后將cDNA第一條鏈與緩沖液、RNase H、dNTPs以及DNA Polymerase I配制成第二鏈合成的反應體系,合成cDNA第二條鏈。cDNA的純化使用QiaQuick PCR試劑盒進行,然后純化后的cDNA做末端修復。將修復后的cDNA 3’末端加上堿基“A”并連接上接頭,隨后對片段的大小進行選擇。最后一步進行PCR反應,構建測序文庫。利用Illumina HiSeq?2000對建好的測序文庫進行測序。將測序獲得的原始數據去除接頭的序列、兩端低質量的序列以及低度復雜的序列,數據的組裝利用Trinity[14]軟件進行。
1.2.3功能注釋首先,通過Blastx程序將Unigene序列與蛋白數據庫NR、Swiss-Prot(Evalue<0.00001)進行比對,并通過Blastn程序將Unigene與核酸數據庫nt(Evalue<0.00001)進行比對,獲得Unigene最高序列相似性的蛋白,便獲得了該Unigene的蛋白質功能注釋信息[14]。然后用Blastx將所得Unigene序列比對到COG(Clusters of Orthologous Groups of Proteins)數據庫(Evalue<0.00001),獲得COG數據庫的功能注釋及其功能分類。Gene Ontology(GO)注釋信息使用軟件Blast2GO[15]獲得,然后使用WEGO軟件[16]對Unigene進行GO功能的分類統計。根據KEGG的注釋信息可進一步得到Unigene的Pathway途徑注釋。
1.2.4香榧SSR位點分析利用MISA軟件(http://pgrc.ipk-gatersleben. de/misa/misa.html)進行SSR位點搜索,搜索參數設置為單、二、三、四、五和六堿基重復。
2 結果與分析
2.1轉錄組測序與組裝
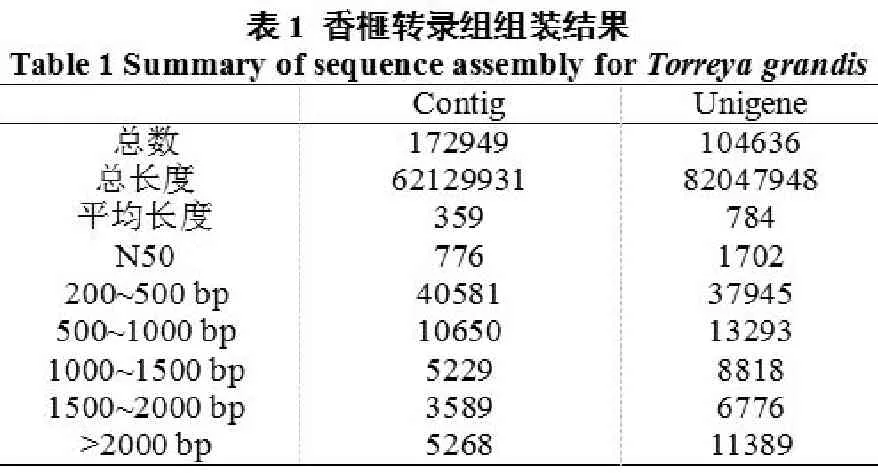
采用Illumina HiSeq?2000對香榧轉錄組進行測序,共得到reads片段37,349,086個,總堿基數約為4.35 G,GC的含量是42.31%,Q20(堿基測序錯誤率小于1%)是96.63%。這些結果說明,轉錄組測序質量良好,獲得的數據可進一步分析。利用組裝軟件Trinity,對測序得到的高品質序列進行de novo從頭組裝,共獲得了172,949個Contigs,平均的長度為359 nt,N50為776。其中,40,581個Contig長度為200~500 bp,占23.46%;10,650個Contig長度為500~1000 bp,占6.16%;14,086個Contig的長度為1,000bp以上,占8.14%(表1)。使用組裝軟件將Contig進一步的組裝,獲得了104,636個Unigene,平均長度為784 nt,N50為1,702。其中,37,945個Unigene長度為200~500 bp,占36.26%;13,293個Unigene長度500~1000 bp,占12.7%;8,818個Unigene長度為1000~1500 bp,占8.43%;6,776個Unigene長度為1000~1500 bp,占6.48%;2000 bp以上的Unigene有11,389個,占10.88%(表1)。N50長度可以用來衡量組裝的完整度,Contig和Unigene的N50分別達到了776和1,702,表明了組裝效果良好。
2.2Unigene的功能注釋
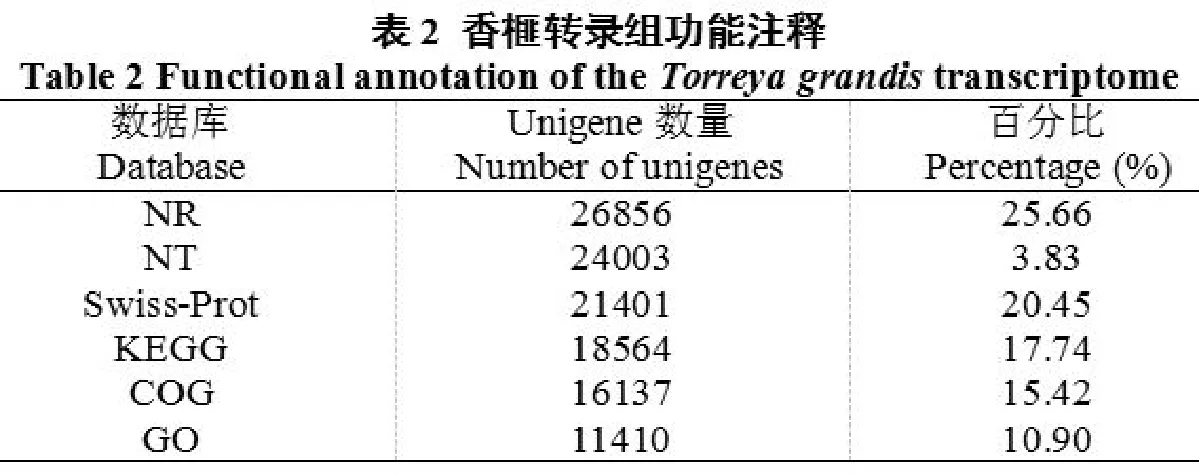
將Unigene序列通過Blast程序比對到數據庫NR,NT,Swiss-Prot,KEGG,COG,GO (Evalue<0.00001),獲得Unigene的功能注釋。在上述數據庫中獲得注釋的Unigenes有28,766個,其中在NR蛋白數據庫中獲得注釋的Unigene有26,856個,在NT數據庫中獲得注釋的Unigenes有24,003個,在Swiss-Prot蛋白數據庫中獲得注釋的Unigene有21,401個(表2)。
2.3香榧轉錄組Unigene的COG功能分類
利用COG數據庫可以對基因產物進行系統進化關系分類。本研究將獲得的Unigene比對到COG數據庫,16,137個Unigene獲得了分類,根據功能可將這些Unigene分為25類(表3),涉及了大多數的生命活動。5,358個Unigene被歸到一般功能預測類,占總數的33.20%。2,959個Unigene被歸到翻譯,核糖體結構及生物發生類,占總數的18.34%。2,654個Unigene被歸到轉錄類,占總數的16.45%。2,106個Unigene被歸到翻譯后修飾,蛋白質折疊及分子伴侶類,占總數的13.05%。2,098個Unigene被歸到信號傳導機制類,占總數的13.00%。而被歸到胞外結構類和核結構類的Unigene最少,分別為10個和3個。
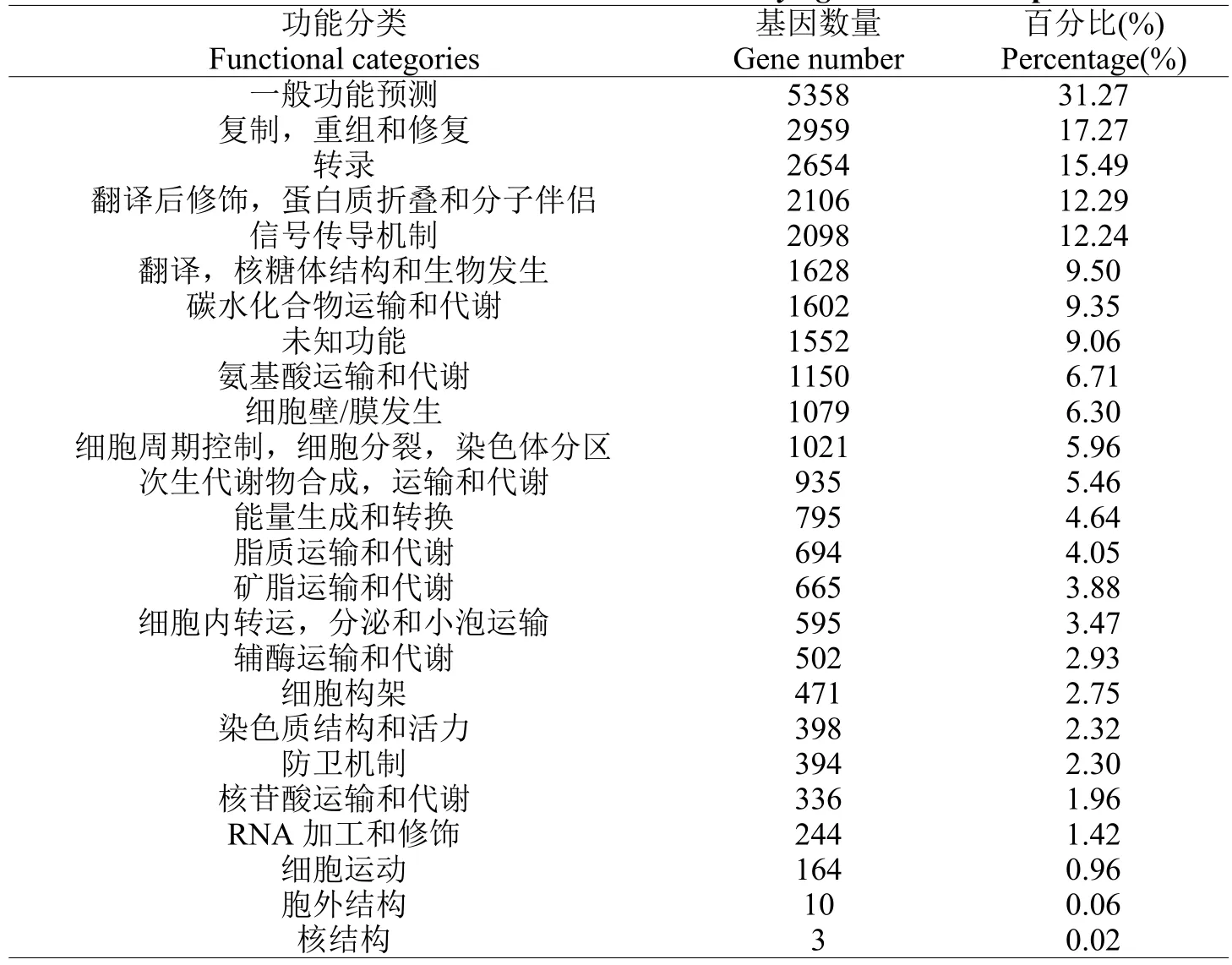
表3 香榧轉錄組的COG分類Table 3 COG functional distribution of the Torreya grandis transcriptome
2.4香榧轉錄組Unigene的GO功能分類
Gene Ontology(GO)基因功能分類是國際上標準化的分類體系之一,生物體中基因與基因產物的屬性由一套標準詞匯表(Controlled vocabulary)來全面描。本研究中11,410個Unigene被歸為55類(圖1)。在參與的生物過程分類中歸類為細胞過程(6,738個),代謝過程(5,684個),單一的生物過程(4,494個),生物調控(2,783個)中的Unigene最多。在所處的細胞位置分類中歸類為細胞(5,260個),細胞器(3,921個),大分子復合物(1,851個),膜(1,388個)中的Unigene最多。在分子功能分類中參與催化活性(6,426個)、結合(5,310個)和轉運活性(380個)的Unigene最多。
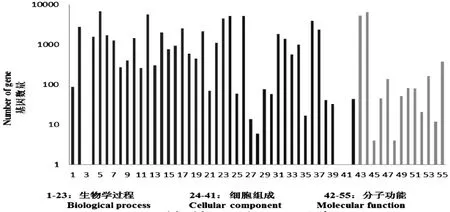
圖1 香榧轉錄組的GO分類Fig.1 Gene ontology classification of the Torreya grandis transcriptome備注:生物學過程:1:生物附著2:生物調控3: cell killing細胞殺傷4:細胞成分和生物合成5:細胞過程6:發育過程7:定位系統建立8:生長;9:免疫系統過程10:定位11:移動12:代謝過程13:多機體過程14:多細胞組織過程15:生物過程的負調控16:生物過程的正調控17:生物過程的調控18:再生19:再生過程20:刺激應答21:律動過程22:信號傳導23:單一的生物過程;細胞組成24:細胞25:細胞連接26:細胞部分27:細胞外基質28:胞外區要素29:胞外區30:胞外區部分31:大分子復合物32:膜33:膜要素34:膜附著腔35:核仁36:細胞器37:細胞器部分38:突觸39:突觸部分40:病毒體41:病毒體部分;分子功能42:抗氧化活性43:結合44:催化活性45:頻道調節器活動46:電子載體活性47:酶調節活性48:通道活性的調節49:分子轉導活性50:核酸結合的轉錄因子的活性51:蛋白結合轉錄因子活性52:刺激應答53:結構分子活性54:翻譯調控因子活性55:轉運活性.Note:Biological process:1: biological adhesion 2: biological regulation 3: cell killing 4: cellular component organization or biogenesis 5: cellular process 6: developmental process 7:establishment of localization 8: growth 9: immune system process 10: localization 11: locomotion 12: metabolic process 13: multi-organism process 14: multicellular organismal process 15: negative regulation of biological process 16: positive regulation of biological process 17: regulation of biological process 18: reproduction 19: reproductive process 20: response to stimulus 21: rhythmic process 22: signaling 23: single-organism process;Cellular component 24: cell 25: cell junction 26: cell part 27: extracellular matrix 28: extracellular matrix part 29: extracellular region 30: extracellular region part 31: macromolecular complex 32: membrane 33: membrane part 34: membrane-enclosed lumen 35: nucleoid 36: organelle 37: organelle part 38: synapse 39: synapse part 40: virion 41: virion part;Molecular function 42: antioxidant activity 43: binding 44: catalytic activity 45: channel regulator activity 46: electron carrier activity 47: enzyme regulator activity 48metallochaperone activity 49: molecular transducer activity 50: nucleic acid binding transcription factor activity 51: protein binding transcription factor activity 52: receptor activity 53: structural molecule activity 54: translation regulator activity 55: transporter activity
2.5香榧轉錄組Unigene的KEGG功能分類
利用KEGG數據庫可以探索基因產物在細胞中的功能及所處的代謝通路,可以用來統計分析基因的產物在生物學上復雜的行為[17]。本次研究中18,564個Unigene被歸到256個小類中。被歸為代謝途徑(Metabolicpathways)的Unigene數量最多,有3,123個,占16.82%。其次是剪接體(Spliceosome),有749個Unigene,占4.03%。第三大類是植物與病原物的互作,有723個Unigene,占3.89%(表4)。
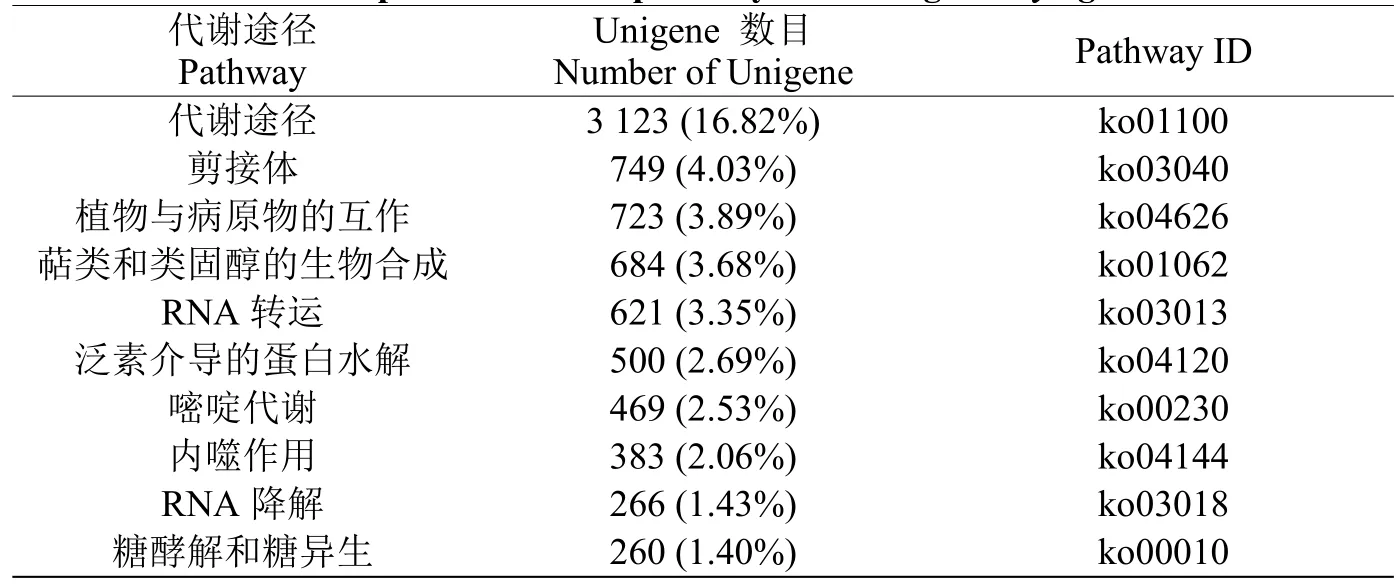
表4 Unigene數量最多的10個代謝通路Table 4 Top ten metabolic pathways involving Torreya grandis
2.6SSR分析
利用軟件MISA搜索香榧Unigene的SSR位點,從104,636個Unigene檢測到了4,706個SSR位點,出現的頻率(檢出的SSR個數和總Unigene的數目之比)為4.5%(表5)。在4,217個Unigene中分布著4,706 個SSR位點,其中414個Unigene含有1個及以上的SSR位點。SSR種類呈現多樣化,單到六核苷酸的重復類型均檢測到。其中位點數量多的SSR類型為三核苷酸,有1,677個;其次為二核苷酸,有1,139個;1,139個SSR位點為單核苷酸;四、五、六核苷酸SSR位點的數量相對較少,分別有69,257,614個。單核苷酸SSR的重復類型主要是A/T(945個);二核苷酸重復的主要類型是AT/AT(507個)、AG/CT (412個)、AC/GT(219個);三核苷酸重復主要類型是AAG/CTT(361個)、AGG/CCT(329個)、AGC/CTG(286個)、AAT/ATT(207個)。對這些SSR的鑒定,有助于開展香榧及其同屬物種的遺傳圖譜構建及基因組差異分析研究。

表5 香榧SSR位點統計結果Table 5 Statistics of SSRs identified in Torreya grandis transcriptome
4 討論
隨著新一代高通量測序技術的不斷進步,植物基因組的測序研究得到了快速的發展,但對香榧基因組的研究非常少。相對于全基因組序列的測定,轉錄組測序技術具有一定的優勢,其一是從頭裝配轉錄本,不需要利用任何以前的基因序列信息;其次測序費用較低,普通的實驗室也能承擔。與其它測序技術相比,Illumina高通量測序技術獲得的數據量大、測序速度快、效率比較高、成本相對較低,已在多種植物上得到應用[18]。本研究利用Illumina測序技術對香榧轉錄組進行了測序,共獲得約4.4 G的原始數據,獲得的數據量比較大,獲得37,349,086個長度為90 bp的序列。利用生物學組裝軟件對這些90 bp的短序列進行了組裝,共獲得Unigene 104,636個。這些研究結果說明Illumina測序技術可以應用在香榧等基因組信息相對匱乏的物種上,并能有效得到轉錄組的信息。
將Unigene序列通過Blast程序與公共數據庫Swiss-Prot、KEGG、NR、NT和COG進行比對,從中獲得注釋的Unigene有28,766個,其中在NR蛋白數據庫中得到注釋的Unigene有26,856個,在Swiss-Prot蛋白數據庫中得到注釋的Unigene有21,401個。104,636個Unigene中只有28,766個在上述的公共數據庫中獲得了注釋信息。造成這樣結果的原因有幾種,一方面是有的Unigene的序列過短,而無法獲得比對信息,另一方面有些Unigene可能是香榧特有的基因。本研究中獲得的Unigene被歸類到多個GO、COG以及KEGG子類中,說明這些組裝的Unigene具有豐富的類型。
SSR分子標記操作相對簡便、具有較好的重復性、獲得的信息量高、覆蓋廣,已經應用在多個方面,包括植物分子輔助育種、遺傳圖譜的構建等方面[19-20]。本研究利用MISA軟件分析了香榧轉錄組,獲得了4,706個SSR位點。其中三核苷酸重復最多,其次是二核苷酸重復和單核苷酸重復,四、五、六核苷酸重復類型的數量較少。根據已有的報道,大多數植物的SSR類型主要以二核苷酸和三核苷酸為主,但是優勢的重復單元也存在差異[21]。本次研究獲得的優勢重復單元是三核苷酸,其次是二核苷酸,這與葡萄(Vitis vinifera)[22]、大麥(Hordeum vulgare L.)[23]等植物一致,比松類高,低于經濟樹種茶。與其親緣關系相近的紅豆杉中以三核苷酸和六核苷酸為主,而且與模式植物擬南芥以三核苷酸為主也相近。水稻中的研究表明相對于三核苷酸和六核苷酸位于基因區而言,二核苷酸和四核苷酸主要位于非編碼區,即基因間區域。從檢出的頻率來看,二核苷酸重復基元中出現最多的是AT/AT、其次是AG/CT和AC/GT,三核苷酸重復中的主要類型是AAG/CTT和AGG/CCT,這些重復基元在大多數雙子葉植物中的出現頻率較高[24]。香榧SSR重復基序的組成與雙子葉植物更加接近,造成這種原因可能與密碼子的偏倚性以及搜索條件的設置有關。由于香榧為非模式植物,可供參考的遺傳信息相對比較少,因此對于其特異性的新基因的挖掘還有待進一步的研究。以本研究獲得的轉錄組為基礎,今后可以開展進一步的開發研究榧樹的分子標記,從而對榧樹的遺傳結構和多樣性展開研究,評估和保護其遺傳資源;還可以克隆榧樹中重要的生物活性成分的合成關鍵基因,以更好地利用其保健、藥用價值和綜合開發利用價值;也為功能基因的挖掘及優良性狀的遺傳改良等提供了大量的遺傳數據資源[25]。
參考文獻
[1]易官美,邱迎君.榧樹的研究現狀與展望[J].資源開發與市場,2013,29(8):844-847
[2]易官美,邱迎君,李曉花,等.榧樹的地理分布與資源調查[J].安徽農業科學,2013,41(19):8200-8202
[3] Sun X,Zhou S,Meng F,et al. De novo assembly and characterization of the garlic(Allium sativum)bud transcriptome by Illumina sequencing[J]. Plant cell reports,2012,31:1823-1828
[4] Seungill K,Myung-Shin K,Yong-Min K,et al. Integrative structural annotation of de novo RNA-Seq provides an accurate reference gene set of the enormous genome of the onion(Allium cepa L.)[J]. DNARes,2015,22(1):19-27
[5] Da Cheng H,Guangbo G,Peigen X,et al. The first insight into the tissue specific taxus transcriptome via Illumina second generation sequencing[J]. PLoS one,2011,6(6):e21220
[6] Miyama M,Tada Y. Transcriptional and physiological study of the response of Burma mangrove(Bruguiera gymnorhiza)to salt and osmotic stress[J]. Plant molecular biology,2008,68(1-2):119-129
[7] Gao C,Wang Y,Liu G,et al. Expression profiling of salinity-alkali stress responses by large-scale expressed sequence tag analysis in Tamarix hispid[J]. Plant molecular biology,2008,66(3):245-258
[8] Mizrachi E,Hefer CA,Ranik M,et al. De novo assembled expressed gene catalog of a fast-growing Eucalyptus tree produced by Illumina mRNA-Seq[J]. BMC genomics,2010,11(6):681
[9] Zhang Y,Zhang S,Han S,et al. Transcriptome profiling and in silico analysis of somatic embryos in Japanese larch (Larix leptolepis)[J]. Plant cell reports,2012,31(9):1637-1657
[10]孫穎,譚曉風,羅敏,等.油桐花芽2個不同發育時期轉錄組分析[J].林業科學,2014,50(5):70-74
[11] Li D,Zhi D,Bi Q,et al. De novo assembly and characterization of bark transcriptome using Illumina sequencing and developmentof EST-SSRmarkersinrubbertree(Heveabrasiliensis Muell.Arg.)[J].BMCgenomics,2012,13(19):1-14
[12] Shi CY,Yang H,Wei CL,et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds[J]. BMC genomics,2011,12(1):1-19
[13]易官美,邱迎君.榧樹居群遺傳多樣性的cpSSR分析[J].果樹學報,2014,31(4):583-588
[14] Grabherr MG,Haas BJ,Yassour M,et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome[J]. Nature biotechnology,2011,29(7):644-652
[15] Conesa A,G?tz S,García-Gómez JM,et al. Blast2GO: a universal tool for annotation,visualization and analysis in functional genomics research[J]. Bioinformatics,2005,21(18):3674-3676
[16] JiaY,LinF,HongkunZ,etal.WEGO:awebtoolforplottingGOannotations[J].Nucleicacidsresearch,2006,34(2):W293-W297
[17] Kanehisa M,Araki M,Goto S,et al. KEGG for linking genomes to life and the environment[J]. Nucleic acids research,2008,36(1):D480-D484
[18] Bie V,Clement L,Reumers J,et al. ViVaMBC: estimating viral sequence variation in complex populations from illumina deep-sequencing data using model-based clustering[J]. BMC bioinformatics,2015,16(1):1-11
[19] Agarwal M,Shrivastava N,Padh H. Advances in molecular marker techniques and their applications in plant sciences[J]. Plant cell reports,2008,27(4):617-631
[20]李炎林,楊星星,張家銀,等.南方紅豆杉轉錄組SSR挖掘及分子標記的研究[J].園藝學報,2014,4(4):735-745
[21] Wei W,Qi X,Wang L,et al. Characterization of the sesame(Sesamum indicum L.)global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers[J]. BMC genomics,2011,12:451
[22] Hong H,Jiang L,Ren Z,et al. Mining and validating grape(Vitis vinifera L.)ESTs to develop EST-SSR markers for genotyping and mapping[J]. Molecular Breeding,2011,28(2):241-254
[23] Varshney RK,Grosse I,H?hnel U,et al. Genetic mapping and BAC assignment of EST-derived SSR markers shows non-uniform distribution of genes in the barley genome[J]. Theoretical and Applied Genetics,2006,113(2):239-250
[24] Chagné D,Chaumeil P,Ramboer A,et al. Cross-species transferability and mapping of genomic and cDNA SSRs in pines[J]. Theoretical and Applied Genetics,2004,109(6):1204-1214
[25]鄧楠,史勝青,常二梅,等.膜果麻黃種子不同發育時期的轉錄組測序分析[J].東北林業大學學報,2015,43(2):28-32
Sequencingand BioinformaticAnalysisfor Transcriptomeof Torreya grandis Fort.ex Lindl.cv.merrillii
YI Guan-mei1,BAO Yan-chun2
1. Ningbo City College of Vocational Technology, Ningbo 315502,China
2. Qingshanhu Landscape Adminstration Office of Nanchang City Jiangxi Province, Nanchang 330039,China
Abstract:Torreya grandis Fort. ex Lindl. cv. Merrillii is an economically important plant on both agriculture and ecology. However,the genomic information of this species has been less studied,leading to limited researching progresses in both molecular biology and gene functions. In the present study,we have sampled different tissues of T. grandi to conduct a transcriptomic analysis using the Illumina HiSeq?2000 technical platform. As a result,a total of 37,349,086 reads were obtained with the whole base number of 4.35 G. Based on the assembling of high quality reads,we identified a total of 104,636 Unigenes with an average length of 784 nt and N50 was 1,702 nt. Comparing these Unigene sequences with those in the public database,28,766 Unigenes were annotated in the Nr database,24,003 Unigenes were in the NT database,and 21,401 Unigenes were in the Swiss-Prot database. Moreover,based on the COG database and the GO database,we also found 16,137 Unigenes and 11,410 Unigenes were in both databases respectively. We further classified 18,564 Unigenes into 256 pathways according to the KEGG annotation information. Finally,we identified 4,706 SSR loci in 4,217 Unigenes via SSR loci searching. The obtained transcriptome data was thus as the first genomic-wide database serving for future studies of T. grandis in terms of functional gene cloning,gene expression,fingerprint construction and molecular marker-assisted breeding.
Keywords:Torreya grandis;transcriptome;Illumina sequencing;SSR
*通訊作者:Author for correspondence. E-mail:yiguanmei@nbcc.cn
作者簡介:易官美(1968-),男,江西進賢人,碩士,副教授.主要從事植物資源學研究. E-mail:875013268@qq.com
基金項目:寧波市科技局農業重大專項項目(2014C11006);寧波市自然科學基金(2015A610267)
收稿日期:2015-05-13修回日期:2015-11-14
中圖法分類號:Q37;S791.53
文獻標識碼:A
文章編號:1000-2324(2016)01-0019-06
