多重PCR靶向富集結合高通量測序篩查結直腸癌中MMR基因的突變
2016-04-21 10:48:55陳慧杰劉方奇
復旦學報(醫學版) 2016年2期
黃 凱 陳慧杰 劉方奇 徐 燁 李 軒 南 蓬
(1復旦大學生命科學學院生物多樣性與生態工程教育部重點實驗室 上海 200433;2上海同達科信生物技術發展有限公司 上海 200080;
3上海生物信息技術研究中心 上海 201203; 4復旦大學附屬腫瘤醫院大腸外科 上海 200032;
5中國科學院上海生命科學研究院植物生理生態研究所 上海 200032)
?
多重PCR靶向富集結合高通量測序篩查結直腸癌中MMR基因的突變
黃凱1,2陳慧杰3劉方奇4徐燁4李軒5南蓬1△
(1復旦大學生命科學學院生物多樣性與生態工程教育部重點實驗室上海200433;2上海同達科信生物技術發展有限公司上海200080;
3上海生物信息技術研究中心上海201203;4復旦大學附屬腫瘤醫院大腸外科上海200032;
5中國科學院上海生命科學研究院植物生理生態研究所上海200032)
【摘要】目的探討多重PCR靶向富集結合高通量測序技術在結直腸癌 (colorectal cancer,CRC)中檢測錯配修復 (mismatch repair,MMR)基因種系突變的應用。方法收集17例CRC患者和14例正常人的血液并提取基因組DNA;設計和優化寡聚核苷酸探針,使其能對5個基因MLHl、MSH2、PMS1、PMS2和MSH6的73個外顯子序列進行有效的PCR擴增和富集;應用多重PCR技術靶向富集樣本MMR基因的外顯子序列;擴增產物進行文庫構建和高通量測序,檢測MLHl、MSH2、PMS1、PMS2和MSH6基因的突變情況。結果31個樣本共得到2.7Gb的數據,平均reads數為287 048,測序數據中平均82.18%可與參考序列進行比對,外顯子序列平均覆蓋度為99.9%,平均測序深度為2 282。在MMR基因的外顯子區域共發現13種非同義單核苷酸變異 (single nucleotide variation,SNV)、2種同義 SNV,其中MSH6的c.G3205C:p.G1069R為未見報道SNV。檢測結果經Sanger法測序驗證,結果一致。結論多重PCR靶向富集結合高通量測序技術是一套通量高、速度快、費用低、準確性高的MMR基因突變篩查方法。
【關鍵詞】錯配修復基因;Lynch綜合征;靶向富集;高通量測序
*This work was supported by the National Key Basic R&D Program of China (973 Plan,2012CB316501),the National Key Technologies R&D Program of China (863 Plan,2012AA02A602) and the National Natural Science Foundation of China (31271409,31401128).
結直腸癌 (colorectal cancer,CRC)是全世界第三大常見惡性腫瘤[1]。研究發現35%的CRC存在家族易感性[2],其中最常見的是Lynch綜合征,即遺傳性非息肉病性結直腸癌 (hereditary nonpolyposis colorectal cancer,HNPCC),占所有病例的2%~5%[3]。錯配修復 (mismatch repair,MMR)基因發生胚系突變是Lynch綜合征的遺傳學發病機制[4-5]。研究發現至少5種MMR基因 (MLH1、MSH2、MSH6、PMS1、PMS2)的胚系突變會導致Lynch綜合征,其中MLH1的胚系突變占50%,MSH2占40%,MSH6占7%,剩下的基因占3%[6]。在家系中,MMR基因種系突變攜帶者為發生Lynch綜合征相關腫瘤的高危人群,檢測MMR基因種系突變能很好地預測Lynch綜合征相關腫瘤的發生危險[7]。
直接基因測序法是檢測MMR基因胚系突變最靈敏和最特異的方法,但Sanger法測序不僅費時而且價格昂貴。除了直接測序外的篩檢方法還有:MMR蛋白表達免疫組化分析 (immunohistoch-meistry,IHC)和微衛星不穩定性 (microsatellite instability,MSI)檢測。IHC檢測可以確定受累基因,從而指導直接測序,不足之處是某些突變只導致MMR蛋白功能受損,而并沒有嚴重影響到MMR的表達,此時IHC檢測可能為陰性[8-10]。另外,有些MSH6胚系突變只導致MMR受損但不會引起MSI[11]。
與傳統的Sanger法測序相比,第二代高通量測序技術具有通量高、速度快、費用低等優點,結合特定核酸序列靶向富集技術,可以實現對疾病最相關的基因進行深度測序。目前已有多種疾病的相關基因運用該技術進行了深度測序,并取得了一定的研究成果[12-14]。本研究對17例CRC病例和14例正常人樣本進行多重PCR靶向富集MMR基因外顯子序列,結合高通量測序技術,檢測5個基因 (MLH1、MSH2、MSH6、PMS1、PMS2)的胚系突變,探討多重PCR靶向富集結合高通量測序在MMR基因胚系突變檢測中的應用,以期為Lynch綜合征的臨床診斷和研究提供新的參考。
資 料 和 方 法
研究資料CRC患者17例,年齡63~81歲,其中男8例,女9例;健康志愿者14例,年齡30~50歲,其中男6例,女8例。CRC樣本由復旦大學附屬腫瘤醫院提供,正常人來自健康志愿者,所有患者及志愿者均知情同意,并簽署了經上海市人類遺傳資源委員會批準的知情同意書。
靶向富集探針的設計使用在線引物設計工具探針 3設計PCR靶向富集MMR基因所需探針,對5個基因的73個外顯子總共11 419個堿基設計了87對探針,PCR擴增產物長度為257~528 bp,該組合探針能覆蓋5個MMR基因 (表1)的所有外顯子區域。引物合成由上海生工生物工程有限公司合成。
MMR相關基因的靶向富集及高通量測序將設計和優化的87對探針分成11組對樣本基因組DNA 進行多重PCR擴增,每組多重PCR的DNA模板量為50 ng,其中第1~8和11組使用多重PCR 5× Master Mix試劑盒(美國NEB公司)進行多重PCR擴增,第9和10組使用GB緩沖液擴增(日本TaKaRa公司LA Tag DNA聚合酶)試劑盒進行多重PCR擴增,擴增條件為:95 ℃預變性1 min;95 ℃變性20 s、63 ℃退火60 s、68 ℃延伸30 s,30個循環;68 ℃延伸5 min。擴增產物經AMpure磁珠(美國Beckman公司)純化后等量混合,取200 ng使用TflexTM快速DNA-Seq試劑盒(美國NEX公司)構建測序文庫,然后用Miseq測序儀進行高通量測序。
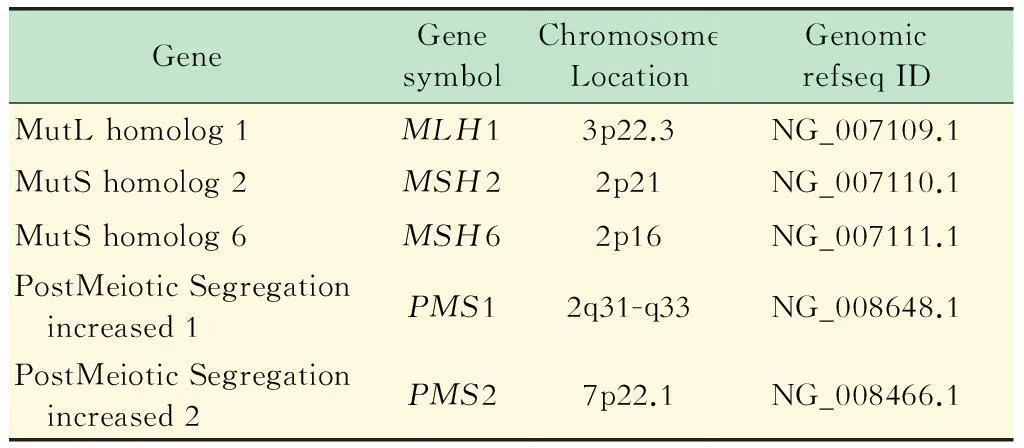
表1 靶向富集探針設計參考序列信息表
生物信息學分析及Sanger測序法驗證測序數據分析使用Bowtie2軟件[15]與人類基因組參考序列 (hg19)進行比對,用SAM工具軟件[16]對結果進行突變篩查分析 (篩選條件:比對質量>20且測序深度>100),用ANNOVAR軟件[17]對篩選的突變進行功能注釋。分析所得結果用Sanger法測序驗證。測序樣本的制備和Sanger測序由上海生工生物工程有限公司完成,測序所用試劑和儀器為BigDye Terminator v1.1 Cycle Sequencing kit 和ABI 3730 測序儀。
結果
靶向富集及測序結果通過多重PCR靶向富集MMR基因和高通量測序,31個樣本共得到2.7 gigabases (Gb)數據,平均每個樣本86 Mb,平均reads數為287 048。其中30個樣本的5個篩查基因 (MLH1、MSH2、MSH6、PMS1、PMS2)外顯子測序覆蓋度為100%,1個為96.8%,平均覆蓋度為99.9%,外顯子區域平均測序深度為2 284。經過生物信息分析,31個測序樣本共發現13種非同義單核苷酸變異 (single nucleotide variation,SNV)、2種同義SNV,在非同義SNV中,位于MLH1基因的有3個:c.A655G:p.I219V、c.T1151A:p.V384D和c.C2101A:p.Q701K;位于MSH2基因的有3個:c.C1168T:p.L390F、c.A1690G:p.T564A和c.G2425A:p.E809K;位于MSH6基因的有3個:c.G116A:p.G39E、c.G3205C:p.G1069R和c.A3488T:p.E1163V;位于PMS2基因的有4個:c.G59A:p.R20Q、c.A1621G:p.K541E、c.C1408T:p.P470S和c.C1454A:p.T485K。2個同義SNV是MSH6基因c.T3306A和PMS2基因c.C780G;基因PMS1未檢測到SNV。這些SNV與數據庫dbSNP和InSight[18](http://www.insight-group.org/mutatioons)進行檢索,發現14種已經報導,其中MSH6的c.G3205C:p.G1069R未見報導 (表2)。
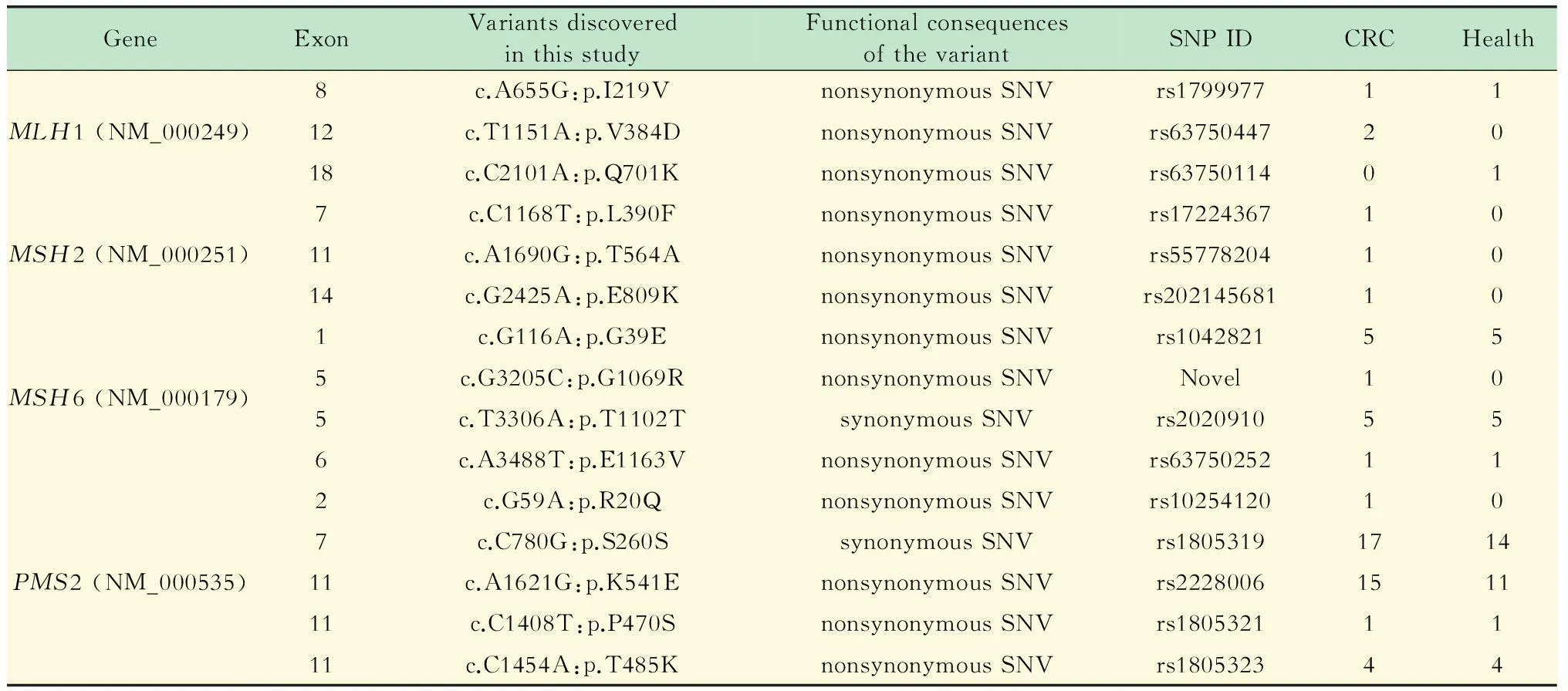
表2 MMR基因突變篩查結果
Sanger測序驗證為了驗證多重PCR靶向富集高通量測序的檢測結果,我們對每種單核苷酸變異類型選取了一個樣本進行了單管的PCR擴增,擴增產物純化后進行Sanger測序。單管PCR擴增及Sanger測序引物與多重PCR靶向富集高通量測序所用的引物相同。結果顯示:Sanger測序驗證結果與多重PCR靶向富集高通量測序結果一致 (圖1)。
討論
目前,新一代測序以其低成本、高通量的優勢成為生命科學研究和臨床疾病基因檢測的重要工具,結合特定核算序列靶向富集技術,可以更加高效經濟的對疾病最相關的基因進行深度測序和研究。Keller等[19]應用靶向富集高通量測序研究多形性成膠質細胞瘤 (glioblastoma multiforme,GBM)SNP,結合全基因組關聯研究 (the genome-wide association study,GWAS)和已知的疾病相關聯SNP,發現一些SNP與吸煙、體質指數、乳腺癌和高分級膠質瘤相關聯。Ying等[20]運用目標區域捕獲高通量測序技術對6個苯丙酮尿癥相關基因 (PAH、PTS、GCH1、QDPR、PCBD1和GFRP)的所有外顯子進行了突變檢測,共發現了PAH基因中23個已知變異,以及6個PAH和PTS的新型突變。
CRC是我國最常見的惡性腫瘤之一,并有逐年上升的趨勢,部分CRC具有家族遺傳易感性。由于我國的計劃生育政策和城市化人口遷徙,家系正變得越來越小,家系成員也越來越趨于分散,這對遺傳性CRC的研究和診斷帶來一定的挑戰。因此,對普通CRC患者或健康人員進行遺傳學檢查,以確認或排除腫瘤遺傳易感性是具有一定應用價值的。MMR基因的胚系突變被認為是最常見的遺傳性CRC-Lynch綜合征發生的分子遺傳學基礎,檢測MMR基因種系突變能很好地預測Lynch綜合征相關腫瘤的發生危險。目前檢測MMR基因突變的方法依然是單個基因逐一檢測且以一代測序為主[21-23],但此方法費時、通量低且價格昂貴,不適合臨床大樣本量的檢測,而高通量測序應用于MMR基因檢測的報道極少。因此,我們設計了一套探針,通過多重PCR靶向富集MMR基因,結合高通量測序檢測MMR基因的胚系突變。
為了能夠有效的多重PCR靶向富集5個MMR基因MLH1、MSH2、MSH6、PMS1、PMS2的外顯子序列,我們設計了87對探針,覆蓋了5個基因73個外顯子共11 419個堿基序列,PCR產物的長度為257~528 bp。設計的探針不僅包含了外顯子區域,也包含了外顯子與內含子的結合位置,同時,這些探針具有相近的TM值,這樣多重PCR時可以使用相同的退火條件。經過實驗優化,我們將87對探針分成11組進行多重PCR,每組包含2~11對探針,其中第9、10組擴增區域GC含量較高,使用TaKaRa LA Tag with GC Buffer試劑盒能有效擴增,其余的多重PCR使用NEB MixMultiplex PCR 5× Master Mix試劑盒進行擴增。多重PCR反應條件經過多次實驗優化后可使每對探針都能夠有效擴增,其擴增條件為:95 ℃預變性1 min;95 ℃變性20 s、63 ℃退火60 s、68 ℃延伸30 s,30個循環;68 ℃延伸5 min。多重PCR產物經過純化后可進行測序文庫制備,在測序文庫構建時加入標簽序列,可以實現多樣本的檢測。本研究使用了31種標簽序列,對17例CRC病例和14CRC正常人的樣本進行了標記。通過引入標簽序列,使得多樣本基因檢測更加高效快捷。以高通量測序平臺Miseq和一代Sanger測序平臺ABI 3730為例,我們將自己設計的多重PCR靶向富集結合高通量測序的方法和傳統的單管PCR-Sanger測序法檢測5個MMR基因外顯子所用時間和費用進行了比較。單管PCR-Sanger測序需對每個基因的外顯子進行單管PCR擴增,擴增產物純化后進行Sanger法測序,每檢測一個樣本的MMR基因胚系突變需要分別進行87個單管PCR及87個Sanger測序反應;多重PCR技術可同時擴增多個MMR基因外顯子序列,擴增產物純化后進行文庫構建并測序,每檢測1個樣本只需11管多重PCR和1次文庫構建。如表3所示:由于受合成引物費用的影響,當檢測少量樣本時,例如檢測1個樣本,所產生的費用主要是引物合成費用;同時,二代測序儀運行通量高,每次運行可同時檢測上百個樣本,當檢測單個樣本時需要和其他樣本混合測序,時間上比一代測序花費更多;但當樣本數量達到100個或更多時,檢測時間和費用主要受限于PCR擴增和測序技術,靶向富集結合高通量測序檢測MMR基因突變的時間和費用要遠遠小于單管PCR-Sanger法測序。因此,多重PCR靶向富集結合高通量測序技術是一套通量高、速度快、費用低的MMR基因突變篩查方法,適合臨床大樣本量的MMR基因突變篩查。
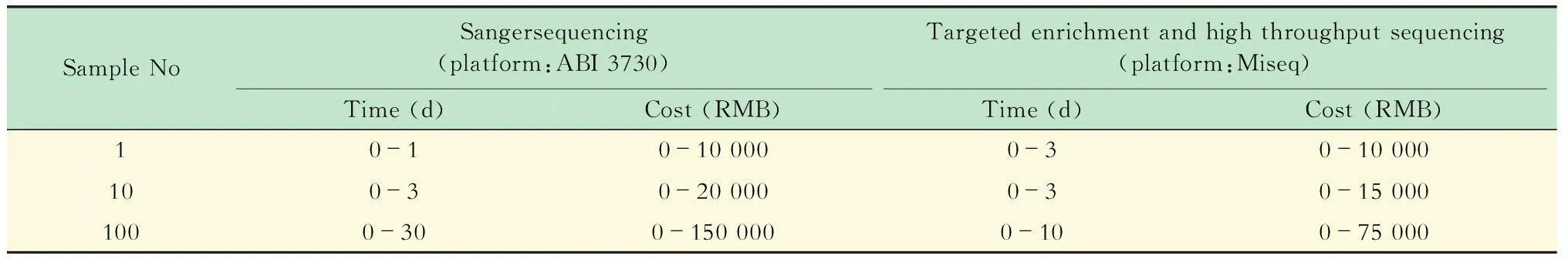
表3 MMR基因外顯子測序費用比較
利用多重PCR靶向富集高通量測序,我們在17例DRC病例和14例正常人的樣本中共發現外顯子區域13種非同義單核苷酸變異、2種同義單核苷酸變異。與dbSNP數據庫進行檢索,發現MSH6的c.G3205C:p.G1069R為未報導的單核苷酸變異,其余14種為已報導單核苷酸變異。將這些已知單核苷酸變異位點進行數據庫ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/)檢索發現:位于MLH1基因第18外顯子c.C2101A:p.Q701K為致病或可能致病的突變;位于MSH2基因第14外顯子c.G2425A:p.E809K為致病性不確定的突變;其余10種非同義單核苷酸變異為可容忍的變異,可能與CRC的發生沒有關聯。致病或可能致病的突變c.C2101A:p.Q701K由范怡梅等[22]于2005年首次發現,并在隨后的功能分析表明此變異造成MLp和PMS2互動效率降低,可能提高突變攜帶者患胃腸腫瘤的風險[24]。在另一項研究中,c.C2101A:p.Q701K在2例腫瘤樣本中檢測出,而在100例非腫瘤對照樣本中并沒有發現該突變,同時對這兩例攜帶該突變的腫瘤樣本進行MSI檢測,5個常規位點檢測出4個陽性結果[25]。本研究在一例健康人 (年齡<50歲)中檢測出該突變,提示該志愿者有可能存在腫瘤遺傳易感性,應詢問是否有患病家族史,并做進一步的檢測 (IHC或者MSI)和隨訪;致病性不確定的突變c.G2425A:p.E809K在1例CRC患者中檢測出,同時檢索1 000 Genomes Projects (2012 release)數據庫發現該等位基因的頻率只有0.000 5,經Mutation Taster預測該單核苷酸變異為有害變異[26],可能會影響MMR的功能,提示該變異可能與CRC的發生有關。在本研究中新發現一種單核苷酸變異,即位于MSH6的第5外顯子c.G3205C:p.G1069R,此變異在1例CRC患者中檢測出,經Mutation Taster預測該單核苷酸變異為有害變異,推測可能與CRC的發生有關。然而,進一步的功能分析研究是必要的,以確認MSH2基因c.G2425A:p.E809K和MSH6基因c.G3205C:p.G1069R的臨床意義。每種類型的非同義SNV我們都挑選了一個樣本進行Sanger測序,結果與高通量測序一致,表明靶向富集高通量測序技術對于發現MMR基因的胚系突變具有高度的準確性。
多重PCR靶向富集結合高通量測序技術用于MMR基因的外顯子突變檢測準確且經濟、高效,其與臨床表型對照研究,可為Lynch綜合征遺傳風險的評估和治療方案的制定提供新的參考。
參考文獻
[1]PETO J.Cancer epidemiology in the last century and the next decade[J].Nature,2001,411 (6835):390-395.
[2]LICHTENSTEIN P,HOLM NV,VERKASALO PK,etal.Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden,Denmark,and Finland[J].NEnglJMed,2000,343 (2):78-85.
[3]SAMOWITZ WS,CURTIN K,LIN HH,etal.The colon cancer burden of genetically defined hereditary nonpolyposis colon cancer[J].Gastroenterology,2001,121 (4):830-838.
[4]JASS JR.Role of the pathologist in the diagnosis of hereditary non-polyposis colorectal cancer[J].DisMarkers,2004,20 (4-5):215-224.
[5]BOLAND CR.Decoding hereditary colorectal cancer[J].NEnglJMed,2006,354 (26):2815-2817.
[7]李曉鷗,盛劍秋,付蕾,等.錯配修復基因突變檢測對遺傳性非息肉病性結直腸癌患病風險的預測[J].中華消化雜志,2009,29 (11):721-725.
[8]ROBINSON KL,LIU T,VANDROVCOVA J,etal.Lynch syndrome (hereditary nonpolyposis colorectal cancer)diagnostics[J].JNatlCancerI,2007,99 (4):291-299.
[9]LYNCH HT,LYNCH JF,LYNCH PM.Toward a consensus in molecular diagnosis of hereditary nonpolyposis colorectal cancer (Lynch syndrome)[J].JNatlCancerI,2007,99 (4):261-263.
[10]RAECAARA TE,KORHONEN MK,LOHI H,etal.Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLp[J].Gastroenterology,2005,129 (2):537-549.
[11]SALOVAARA R,LOUKOLA A,KRISTO P,etal.Population-based molecular detection of hereditary nonpolyposis colorectal cancer[J].JClinOncol,2000,18 (11):2193-2200.
[12]GERRARD G,VALGANON M,FOONG HE,etal.Target enrichment and high-throughput sequencing of 80 ribosomal protein genes to identify mutations associated with Diamond-Blackfan anaemia[J].BrJHaematol,2013,162 (4):530-536.
[13]SCOTT CA,PLAGNOL V,NITOIU D,etal.Targeted sequence capture and high-throughput sequencing in the molecular diagnosis of ichthyosis and other skin diseases[J].JInvestDermatol,2013,133 (2):573-576.
[14]LIN HH,SINNER MF,BRODY JA,etal.Targeted sequencing in candidate genes for atrial fibrillation:the cohorts for heart and aging research in genomic epidemiology (CHARGE) targeted sequencing study[J].HeartRhythm,2014,11 (3):452-457.
[15]LANGMEAD B,SALZBERG S.Fast gapped-read alignment with Bowtie 2[J].NatMethods,2012,9 (4):357-359.
[16]LI H,HANDSAKER B,WYSOKER A,etal.The Sequence Alignment/Map format and SAM tools[J].Bioinformatics,2009,25 (16):2078-2079.
[17]WANG K,LI MY,HAKONARSON H.ANNOVAR:Functional annotation of genetic variants from next-generation sequencing data[J].NucleicAcidsRes,2010,38 (16):e164.
[18]KOHONEN-CORISH MR,MACRAE F,GENUARDI M,etal.Deciphering the colon cancer genes-report of the InSiGHT-Human Variome Project Workshop,UNESCO,Paris 2010[J].HumanMutat,2011,32 (4):491-494.
[19]KELLER A,HARZ C,MATZAS M,etal.Identification of novel SNPs in glioblastoma using targeted resequencing[J].PLoSOne,2011,6 (6):e18158.
[20]GU Y,LU KM,YANG GH,etal.Mutation spectrum of six genes in Chinese phenylketonuria patients obtained through next-generation sequencing[J].PLoSOne,2014,9 (4):e94100.
[21]WEI WQ,LIU FQ,LIU L,etal.Distinct mutations in MLp and MSh1 genes in hereditary non-polyposis colorectal cancer (HNPCC) families from china[J].BMBRep,2011,44 (5):317-322.
[22]FAN YM,LIU XR,ZHANG H,etal.Variations in exon7 of the MSh1 gene and susceptibility to gastrointestinal cancer in a Chinese population[J].CancerGenetCytogenet,2006,170 (2):121-128.
[23]KIM YM,CHOE CG,CHO SK,etal.Three novel germline mutations in MLp and MSh1 in families with Lynch syndrome living on Jeju island,Korea[J].BMBRep,2010,43 (10):693-697.
[24]FAN YM,WANG W,ZHU M,etal.Analysis of hMLp missense mutations in East Asian patients with suspected hereditary nonpolyposis colorectal cancer.[J].ClinCancerRes,2007,13 (24):7515-7521.
[25]YAP HL,CHIENG WS,LIM RC,etal.Recurring MLp deleterious mutations in unrelated Chinese Lynch syndrome families in Singapore[J].FamCancer,2009,8 (2):85-94.
[26]SCHWARZ JM,RODELSPERGER C,SCHUELKE M,etal.Mutation Taster evaluates disease-causing potential of sequence alterations[J].NatMethods,2010,7 (8):575-576.
Distinct mutations ofMMRgene in colorectal cancer by targeted enrichment and high-throughput next generation sequencing
HUANG Kai1,2,CHEN Hui-jie3,LIU Fang-qi4,XU Ye4,LI Xuan5,NAN Peng1△
(1MinistryofEducationKeyLaboratoryforBiodiversityScienceandEcologicalEngineering,SchoolofLifeSciences,FudanUniversity,Shanghai200433,China;2Tongji-SCBITBiotechnologyCo.,Ltd.,Shanghai200080,China;3ShanghaiCenterforBioinformationTechnology,Shanghai201203,China;4DepartmentofColorectalSurgery,ShanghaiCancerCenter,FudanUniversity,Shanghai200032,China;5InstituteofPlantPhysiologyandEcology,ShanghaiInstitutesforBiologicalSciences,ChineseAcademyofSciences,Shanghai200032,China)
【Abstract】ObjectiveTo detect the germline mutations of mismatch repair (MMR) genes in colorectal cancer (CRC) using targeted enrichment and high-throughput next generation sequencing,and to explore its applications in research and clinical diagnosis of Lynch syndrome.MethodsGenomic DNA was extracted from 17 patients diagnosed with colorectal cancer and 14 healthy adults, Primers,which could amplify all the 73 exons and flanking regions of 5 MMR genes (MLH1,
MSH2,MSH6,PMS1,PMS2) by multiplex PCR were designed and optimized,PCR products were then sequenced by Illumina Miseq sequencer.ResultsWe obtained approximately 2.7 giga-base sequence data,and the average reads number of individual samples was 287048.On average,82.18% of the reads could be mapped to the reference human genome (HG19).Average coverage and sequencing depth of targeted regions were 99.9% and 2282-fold respectively.After bioinformatic analysis,we found 14 previously annotated single-nucleotide variants (SNVs) in 5 mismatch repair (MMR) genes and 1 novel mutations inMSH6 genes (c.G3205C:p.G1069R).These results were confirmed by sanger sequencing.ConclusionsTargeted enrichment combined with high-throughput next generation sequencing can be used to detect mutations in MMR genes with high sensitivity and lower cost than conventional methods.
【Key words】mismatch repair gene;lynch syndrome;targeted enrichment;high-throughput sequencing
(收稿日期:2015-07-24;編輯:段佳)
【中圖分類號】R735.3,Q781
【文獻標識碼】A
doi:10.3969/j.issn.1672-8467.2016.02.014
國家重點基礎研究發展計劃(973計劃)項目 (2012CB316501);國家高技術研究發展計劃(863計劃)項目 (2012AA02A602);國家自然科學基金 (31271409,31401128)
△Corresponding authorE-mail:nanpeng@fudon.edu.cn
