高電壓正極材料Li1-xCoO2(x=0.75) 的電子結構
2016-06-22 06:48:42胡家琦朱梓忠
廈門大學學報(自然科學版) 2016年3期
林 妹,胡家琦,朱梓忠
(廈門大學物理科學與技術學院,福建廈門361005)
高電壓正極材料Li1-xCoO2(x=0.75) 的電子結構
林妹,胡家琦,朱梓忠*
(廈門大學物理科學與技術學院,福建廈門361005)
摘要:進一步提高目前成熟的鋰離子電池電極材料LiCoO2的容量是一個重要的研究方向.本文采用基于密度泛函理論的第一性原理方法研究了高脫鋰量材料Li1-xCoO2(x=0.75)的電子結構和晶體結構性質,為深入理解該高電壓材料的電子結構提供基礎.計算表明,LiCoO2材料中Co是以正常的價態+3價的形式存在的.少量脫鋰時,小部分Co3+由于進一步失去電子而從+3價變成+4價.當深度脫鋰(x=0.75)時,不僅有很多的Co3+再變價,而且部分O-2p軌道也會失去電子,產生2種氧離子O1和O2,其中O1離子占O變價總數的1/3,而O2離子占2/3.不同價態的Co與不同氧離子之間的鍵長有明顯的區別.與Co3+相比,Co4+和氧離子之間聚集了更多的電子,它們之間的相互作用力也將強于Co3+與氧離子之間的相互作用力.
關鍵詞:Li0.25CoO2;電子結構;第一原理計算;GGA+U
鋰離子電池具有比能量高、自放電小和壽命長等優點,是電動汽車最具實用可能性的動力電池.電動汽車對鋰離子電池的能量密度提出了巨大的挑戰.鋰離子電池正極材料LiCoO2的首次出現可追溯到20世紀50年代中期[1],它也是目前廣泛商業化應用的第一代鋰離子電池正極材料,在鋰離子電池產業中的電子設備和電動車領域應用最為廣泛[2-3],在過去30多年間得到了廣泛的研究.LiCoO2中所有的Li+從材料中完全脫出時,它的理論容量達到274 mAh/g,當電壓在3.0~4.3 V (vs.Li/Li+) 時實際容量可達到160 mAh/g,這時充放電過程中每個分子式LiCoO2單元大約有0.55個Li+被脫出[4].當充放電過程中每個分子式的LiCoO2單元有0.65~0.70個Li+被脫出時,大約對應于4.5 V的電壓.所以,當原子分數x=0.75 時,對應的Li1-xCoO2是一個高電壓的正極材料,它是目前LiCoO2正極材料領域非常重要的研究方向之一,同時也是進一步提高電池能量密度的重要途徑之一.
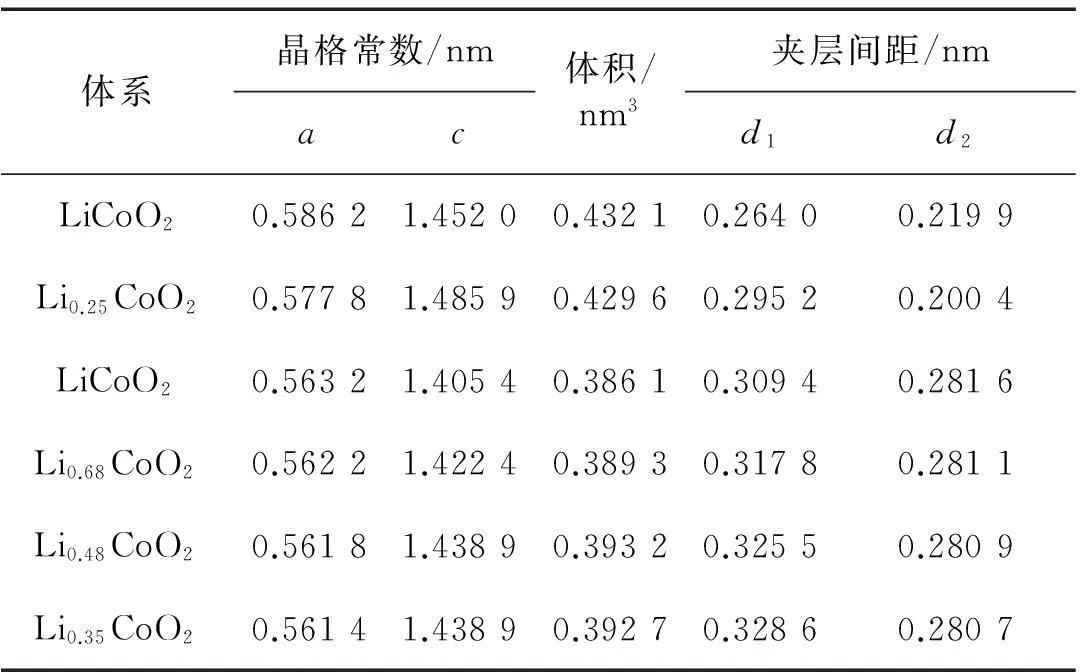
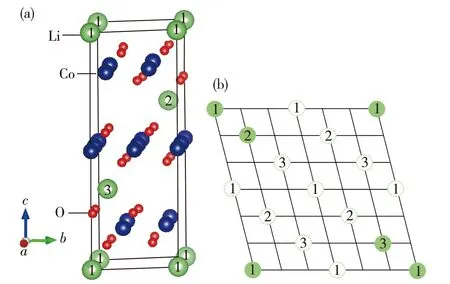
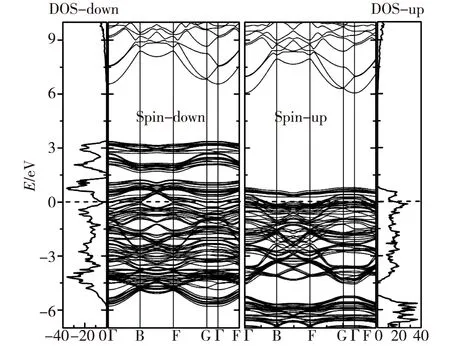
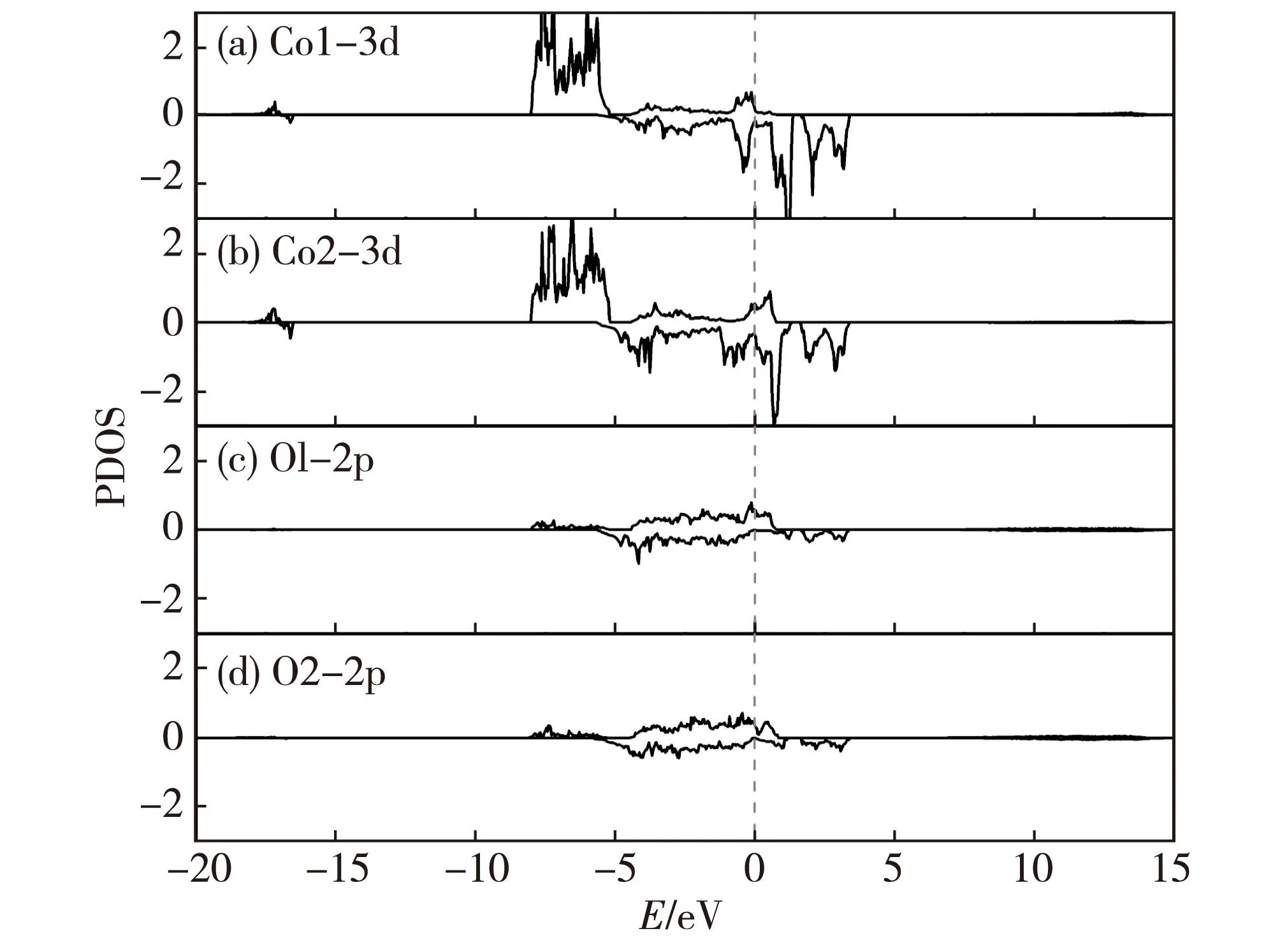
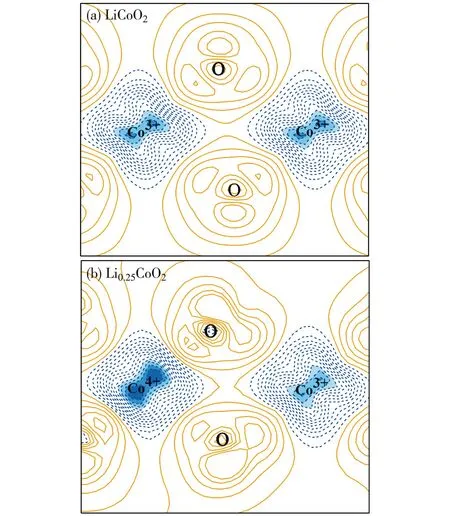
過去對未脫鋰相LiCoO2和脫鋰相Li1-xCoO2的晶體、電子結構和物理化學性質的理論分析和實驗探索用了許多不同的研究方法.這里,我們重點討論LiCoO2在脫鋰過程中結構相變的情況(涉及到多種相之間的轉化).Milewska等[5]通過理論和實驗研究了Li1-xCoO2脫鋰體系在0 本文中,我們采用第一性原理的理論方法,對深度脫鋰的Li0.25CoO2高電壓鋰離子電池正極材料的幾何結構進行了計算,在獲得原子結構的基礎上進行了電子能帶結構、電子態密度和電荷密度性質等比較全面的計算,為深入理解該高電壓材料的電子結構提供了理論基礎. 1計算方法 本文的計算采用基于密度泛函的第一性原理方法,使用的程序包是Viennaabinitiosimulation package(VASP)[12-13].該程序包采用平面波展開、映射綴加波勢表述(PAW)[14]以及廣義梯度近似(GGA)[15]或局域密度近似(LDA)形式的交換關聯勢.眾所周知,使用GGA的計算對插層復合物材料結構性能的預測比較準確,但會很大程度上低估材料的帶隙寬度和電壓值.所以,經常需要通過GGA+U的處理方法來改善GGA計算所帶來的不足,進而比較準確地計算出插層復合物材料的帶隙寬度和電壓.考慮到已有報道的比較好的U值[16-17],我們把Co原子的赫伯德Ueff值取為 5 eV,通過計算表明它比較適合于Li1-xCoO2體系.此外,計算時平面波動能的截斷取為500 eV,布里淵區的積分采用Monkhorst-Pack[18]的特殊k點取樣方法,電子結構的計算均采用5×5×2的k網格點. 對于層狀材料Li1-xCoO2(x=0.75)的幾何結構和電子結構的計算是在2×2×1的超原胞中進行的,它包含了12個分子式單元,即單位原胞包含了12個Li,12個Co和24個O原子(當x=0.75時,只含3個Li+).在進行電子結構的計算前,晶格常數和所有的原子位置均進行了充分的弛豫,直到各原子上的受力收斂于0.1 eV/nm.由于Co原子的磁性可能對材料電子結構的性質有重要影響,所以所有的計算都是在自旋極化的密度泛函理論框架下進行的. 2結果和討論 2.1高電壓脫鋰態Li1-xCoO2(x=0.75)的晶體結構 確定Li+位置的具體做法如下:1) 利用Wu等[19]開發的一種自適應遺傳算法(AGA)擬合得到關于Li-Co-O體系的嵌入原子勢(EAM);2) 利用大量的隨機數來產生大量的隨機結構(每個原子的位置用3個隨機數描述);3) 在使用EAM的情況下計算各隨機結構的能量.由于EAM方法計算能量的速度非常快,所以可以篩選大量的由隨機數產生的結構(例如:這里的1 000萬個隨機數);4) 去除大量的高能量的隨機結構樣本.選取適當數目的低能量結構,繼續在EAM框架下對各個結構的原子位置進行弛豫;5) 在結構弛豫的基礎上,挑選一些結構做密度泛函理論下的第一原理計算(含原子弛豫),能量最低的結構即被認為是最終的晶體結構. 表1給出了弛豫后的LiCoO2和Li1-xCoO2材料的結構參數以及實驗值.計算所得的原胞體積比實驗值稍大,而原子層間距比實驗值略小.整體上,理論計算值和實驗數據比較符合.當Li+從正極材料脫出時,晶格常數a變化不大,而c增大較明顯,這是由于帶正電的Li+層和帶負電的CoO6層之間存在較弱的庫侖相互作用.可以看到,脫鋰中O—O層間距持續發生變化.Li+層兩邊的O—O層間距從LiCoO2中的0.264 0 nm變化為Li0.25CoO2中的0.295 2 nm,表明該O—O層之間的相互作用持續在變弱.另一方面,Co層兩邊的O—O層間距從LiCoO2中0.219 9 nm變為Li0.25CoO2的0.200 4 nm,說明Co—O的離子相互作用在增強,這樣的結構特征和理論結果是相符的[20].使用上述方法“弛豫”后得到的Li0.25CoO2材料的Li+最優化結構,如圖1所示.滿鋰態LiCoO2中有12個Li+的位置,在Li0.25CoO2中,12個位置只有3個被Li+所占據,它們分別均勻分布于超原胞的每一層,而且由于Li+間的排斥作用而使得不同層的Li+錯開排列. 表1 Li1-xCoO2的結構參數 注:d1和d2分別標記Li層和Co層兩邊的O—O層間距. 數字表示Li+所在的原子層;實心綠色圓為被Li+所占據的位置.圖1 Li0.25CoO2的晶體結構和Li+的位置(a)及 Li+的位置在(011)面上的投影(b)Fig.1Structure of Li0.25CoO2 and the positions of Li+(a) and the projection of Li+ positions onto the (011) plane(b) 2.2高電壓脫鋰態Li1-xCoO2(x=0.75)的電子結構 滿鋰態下的LiCoO2為半導體,其計算所得帶隙寬度約為1.34 eV.費米能級附近主要是Co-3d態和O-2p態的貢獻,能量范圍在-5~0 eV之間的Co-3d和O-2p軌道具有較強的雜化作用,在2~4 eV之間也有部分的雜化.從Li0.25CoO2的能帶圖和相應的總態密度圖中(如圖2所示)可以清晰地看到有能帶穿越費米能級,帶隙消失,使得深度脫鋰態的Li0.25CoO2材料具有金屬性質.另外,自旋向上的能帶與自旋向下的能帶有明顯的差別,說明該材料有明顯的自旋極化.滿鋰態的LiCoO2體系中,Co的正常價態是Co3+,然而在脫鋰的Li1-xCoO2體系中,Co的平均價態將比+3價高,也即隨著一部分鋰的脫出,部分Co3+將繼續失去1個電子變成Co4+.Li0.25CoO2的分波態密度如圖3(a)所示表示1個Co3+,而圖3(b) 表示因失去1個電子形成的Co4+.在深度脫鋰Li0.25CoO2的情況下,不僅有Co3+和Co4+的氧化反應,而且O也會失去電子產生2種氧離子O1和O2,參見圖3(c)和(d),這種現象與Li2FeSiO4中的氧離子在深度脫鋰時會被氧化的情況一致[21].在這里,超原胞中包含有24個氧離子,其中O1離子占1/3,而O2離子占2/3.O1和O2離子最大的不同在于費米能級處的態密度分布,O1離子的態密度在費米能級處有值,而O2離子則為零.從Li0.25CoO2的結構中發現:Co4+周圍是由4個O1離子和2個O2離子形成的八面體,Co3+的周圍是由4個O2離子和2個O1離子形成的八面體.此外,Co4+與O2之間的鍵長比Co4+和O1之間的鍵長要長,而 Co3+的情況則相反,即Co3+與O2之間鍵長比Co3+與O1之間的鍵長要短. 圖2 Li0.25CoO2的能帶結構和總態密度圖Fig.2Band structures and total density of states (DOS) for Li0.25CoO2 system 圖3 Li0.25CoO2的分態密度圖Fig.3Partial density of states (PDOS) for the Li0.25CoO2 system 圖4給出了LiCoO2和Li0.25CoO2的差分電荷密度.可以發現,Co—O之間所成的鍵有明顯單鍵的離子性,同時共價性也有著重要作用.這可以從圖4中的差分電荷密度看到:O周圍有電子聚集(獲得電子),而Co周圍則失去電子.同時,O周圍和Co周圍的電子分布有明顯的方向性.從兩圖的對比可以得出,由于氧離子更靠近Co4+,Co4+與氧離子之間的共價性更強,而且Co4+和氧離子之間聚集了更多的電子,這些特征使得Co4+與氧離子之間的相互作用力強于Co3+與氧離子之間的相互作用力. 等高線的范圍:-0.75~0.20 e/10-3nm3;等高線間隔:0.05 e/10-3nm3;其中藍色虛線表示失去電子的區域,橘紅色實線代表得到電子的區域.圖4 Li1-xCoO2 (x=0 和0.75) 的差分電荷密度圖Fig.4Contour plots of deformation charge densities for Li1-xCoO2 (x=0 and 0.75) 3結論 本文使用基于密度泛函理論的第一性原理方法計算了深度脫鋰態Li1-xCoO2(x=0.75)材料的晶體結構和電子結構性質.計算結果表明,深度脫鋰態的Li1-xCoO2(x=0.75)材料由于有能帶穿越費米能級而呈現金屬性,而且能帶有明顯的自旋極化的特征.該高脫鋰量(x=0.75)體系仍然保持了層狀結構.未脫鋰的LiCoO2材料中Co是以正常的價態+3價的形式存在的.少量脫鋰時,相應的小部分Co3+會進一步失去電子而從+3價變成+4價.當深度脫鋰(x=0.75)時,不僅有很多的Co3+再變價,而且部分O-2p軌道也會失去電子,產生2種氧離子O1和O2,其中O1離子占O變價總數的1/3,而O2離子占2/3.此外,與Co3+—O相比,Co4+—O之間聚集了更多的電子,它們之間的相互作用也強于Co3+與氧離子之間的相互作用.這種相互作用也從不同價態的Co與不同狀態的氧離子之間的鍵長的明顯差別中體現出來.為了進一步提高LiCoO2的能量密度,重要途經之一是通過提高正極材料的電壓來實現,這必須通過在LiCoO2材料中脫出更多的鋰離子來達到目的.目前主要的問題是鋰電池的循環性能不好,當超過50%的鋰脫出時,其結構會不穩定和發生相變.本文得到的Li1-xCoO2(x=0.75)的電子結構和晶體結構的信息,有助于人們理解高電壓正極材料Li1-xCoO2的物理和電化學性質,也為制備高能量密度的LiCoO2為正極材料的鋰電池提供了理論幫助. 參考文獻: [1]JOHNSTON W D,MIUER R C.A study of several systems of the type Lix[CoyNi(1-y)]O[J].J Phys Chem,1958,63:198-202. [2]SCROSATI B,GARCHE J.Lithium batteries:status,prospects and future[J].Journal of Power Sources,2010,195:2419-2430. [3]KOKSBANG R,BARKER J,SHI H,et al.Cathode materials for lithium rocking chair batteries[J].Solid State Ionics,1996,84:1-21. [4]WANG Z G,WANG Z X,PENG W J,et al.Structure and electrochemical performance of LiCoO2cathode material in different voltage ranges[J].Ionics,2014,20:1525-1534. [6]IMANISHI N,FUJIYOSHI M,TAKEDA Y,et al.Preparation and Li-NMR study of chemically delithiated Li1-xCoO2(0 [7]HONG J S,SELMAN J R.Relationship between calorimetric and structural characteristics of lithium-ion cells[J].J Electrochem Soc,2000,147(9):3183-3189. [8]VAN D V,AYDINOL M K,CEDER G.First-principles evidence for stage ordering in LiCoO2[J].J Electrochem Soc,1998,145(6):2149-2155. [9]CHEN Z,DAHN J R.Methods to obtain excellent capacity retention in LiCoO2cycled to 4.5 V[J].J Electrochem Soc,2004,A49:1079-1090. [10]CHEN Z,LU Z,DAHN J R.Staging phase transitions in LixCoO2[J].J Electrochem Soc,2002,149 (12):A1604-A1609. [11]CHANG K K,BENGT H,DENUIS M,et al.Thermodynamic description of the layerd O3 and O2 structural LiCoO2-CoO2pseudo-binary systems[J].Computer Coupling of Phase Diagrams and Themochemistry,2013,41:6-15. [12]KRESSE G,FURTHMüLLER J.Efficiency ofab-initiototal energy calculations for metals and semiconductors using a plane-wave basis set[J].Comput Mater Sci,1996,6(1):15-50. [13]KRESSE G,FURTHMüLLER J.Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J].Phys Rev B,1996,54 (16):169-186. [14]KRESSE G,JOUBERT D.From ultrasoft pseudopotentials to the projector augmented-wave method[J].Phys Rev B,1999,59(3):1759-1775. [15]JOHN P P,KIERON B,MATTHIAS E.Generalized gradient approximation made simple[J].Phys Rev Lett,1996,77(18):3865-3868. [16]ZHOU F,COCOCCIONI M,MARIANETTI C A,et al.First-principles prediction of redox potentials in transition-metal compounds with LDA+U[J].Phys Rev B,2004,70:235121. [17]SOLOVYEV I V,DEDERICHS P H,ANISIMOV V I.Corrected atomic limit in the local-density approximation and the electronic structure of d impurities in Rb[J].Phys Rev B,1994,50(23):16861-16871. [18]MONKHORST H J,PACK J D.Special points for Brillouin-zone integrations[J].Phys Rev B,1976,13 (12):5188-5192. [19]WU S Q,JI M,WANG C Z,et al.An adaptive genetic algorithm for crystal structure prediction[J].J Phys:Condens Matter,2014,26:035402. [20]XIONG F,YAN H J,CHEN Y,et al.The atomic and electronic structure changes upon delithiation of LiCoO2:from first principles calculations[J].Int J Electrochem Sci,2012,7:9390-9400. [21]MASESE T,TASSEL C,ORIKASA Y,et al.Crystal structural changes and charge compensation mechanism during two lithium extraction/insertion between Li2FeSiO4and FeSiO4[J].J Phys Chem C,2015,119:10206-10211. Electronic Structures of High Voltage Cathode Material of Li1-xCoO2(x=0.75) LIN Mei,HU Jiaqi,ZHU Zizhong* (College of Physical Science and Technology,Xiamen University,Xiamen 361005,China) Abstract:To further increase the capacity of the well-developed cathode material LiCoO2 for lithium ion batteries is an important direction of electronic-structure research.In this paper,first-principle calculations based on the density functional theory has been performed to investigate the geometric and electronic structures of Li1-xCoO2(x=0.75)with high amount of deintercalation of Li,in the purpose of providing information for understanding the nature of electronic structures of this high-voltage material.Calculations show that Co is valence +3 in material LiCoO2.When a small amount of Li is extracted from LiCoO2,a portion of Co3+is transformed to Co4+owing to the loss of an electron.When a large amount of Li (x=0.75) is extracted,however,part of electrons of O-2p orbital are also lost,except for the valence change from Co3+to Co4+.Therefore,two different oxidation states of oxygen,O1 and O2,appear.The O1 oxygen takes 1/3 of the total oxidized oxygen,while O2 oxygen takes up the remaining 2/3.There are obvious differences in bond lengths between different valence states of Co and different types of oxygen.More electron accumulation between Co4+and O ions are found,as compared with those between Co3+and O ions.Interactions between Co4+and O ions are also stronger than those between Co3+and O ions. Key words:Li0.25CoO2;electronic structures;first-principle calculations;GGA+U doi:10.6043/j.issn.0438-0479.2016.03.012 收稿日期:2015-07-12錄用日期:2015-08-26 基金項目:國家自然科學基金重點項目(21233004) *通信作者:zzhu@xmu.edu.cn 中圖分類號:O 469 文獻標志碼:A 文章編號:0438-0479(2016)03-0371-05 引文格式:林妹,胡家琦,朱梓忠.高電壓正極材料Li1-xCoO2(x=0.75) 的電子結構.廈門大學學報(自然科學版),2016,55(3):371-375. Citation:LIN M,HU J Q,ZHU Z Z.Electronic structures of high voltage cathode material of Li1-xCoO2(x=0.75).Journal of Xiamen University(Natural Science),2016,55(3):371-375.(in Chinese)
 猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34中學生數理化·七年級數學人教版(2020年11期)2020-12-14 06:59:52中華詩詞(2019年7期)2019-11-25 01:43:04模具制造(2019年3期)2019-06-06 02:10:54藝術品鑒證.中國藝術金融(2018年8期)2019-01-14 01:14:28藝術品鑒證.中國藝術金融(2018年10期)2019-01-08 02:44:26藝術品鑒證.中國藝術金融(2018年6期)2019-01-08 02:43:04藝術品鑒證.中國藝術金融(2018年12期)2018-08-26 06:03:48影視與戲劇評論(2016年0期)2016-11-23 05:26:01新聞傳播(2015年10期)2015-07-18 11:05:40
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34中學生數理化·七年級數學人教版(2020年11期)2020-12-14 06:59:52中華詩詞(2019年7期)2019-11-25 01:43:04模具制造(2019年3期)2019-06-06 02:10:54藝術品鑒證.中國藝術金融(2018年8期)2019-01-14 01:14:28藝術品鑒證.中國藝術金融(2018年10期)2019-01-08 02:44:26藝術品鑒證.中國藝術金融(2018年6期)2019-01-08 02:43:04藝術品鑒證.中國藝術金融(2018年12期)2018-08-26 06:03:48影視與戲劇評論(2016年0期)2016-11-23 05:26:01新聞傳播(2015年10期)2015-07-18 11:05:40
