減肥類保健食品中蒽醌類成分及非法添加化學藥物的測定
2016-09-10 02:14:14錢葉飛陳麗波
食品工業科技 2016年10期
關鍵詞:蘆薈
魯 輝,錢葉飛,張 斌,郝 剛,陳麗波
(蘇州市食品藥品檢驗所,江蘇蘇州 215104)
?
減肥類保健食品中蒽醌類成分及非法添加化學藥物的測定
魯輝,錢葉飛,張斌,郝剛,陳麗波*
(蘇州市食品藥品檢驗所,江蘇蘇州 215104)
建立同時測定減肥類保健食品中非法添加化學藥物氟西汀、比沙可啶以及蒽醌類成分番瀉苷A、番瀉苷B、大黃素、大黃素甲醚、大黃酸、大黃酚、蘆薈苷和蘆薈大黃素的高效液相色譜-二極管陣列檢測器(HPLC-DAD)方法。采用Diamonsil C18色譜柱(250 mm×4.6 mm,5 μm),以乙腈(A)-0.02 mol/L乙酸銨溶液(含0.4%乙酸)(B)為流動相,梯度洗脫(0~5 min,85% B;5~25 min,85 %B~35% B;25~50 min,35% B;50~55 min,35% B~85% B),測定波長為268 nm,流速1.0 mL/min,柱溫25 ℃。對HPLC-DAD檢出氟西汀和比沙可啶的樣品進行電噴霧飛行時間質譜(ESI-Q-TOF/MS)一級掃描及二級碎片離子比對進行陽性確證。結果表明:10種待測物線性關系良好(r≥0.995),平均回收率位于90.3%~104.7%之間,RSD均小于5.0%。實際樣品測定發現25種減肥類保健食品中有2種檢出氟西汀,1種檢出比沙可啶,另有1種樣品同時檢出蘆薈苷和蘆薈大黃素。本文建立的方法具有較高的選擇性和靈敏度,方法分離效果好,耐用性較強,能夠用于減肥類保健食品中這10種成分的定性、定量測定。
HPLC-DAD,減肥類保健食品,非法添加,氟西汀,比沙可啶,蒽醌
近年來,肥胖問題日益突出,人們對減肥類保健食品的需求也越來越大。然而,一些不法廠商為了增強功效,竟非法向減肥類保健食品中添加化學藥物。由于添加的藥物種類和數量往往很隨意,某種程度上將嚴重危害消費者的身體健康[1-2]。為此,國家制定了減肥類保健食品中非法添加酚酞、芬氟拉明、麻黃堿、咖啡因、呋塞米、西布曲明、N-單去甲基西布曲明及N,N-雙去甲基西布曲明等8個化學藥物的檢測標準[3-5]。然而在實際監督檢驗時常發現某些減肥類保健食品中存在含量較高的標準外成分。此類成分一方面可能是商家非法添加的國家標準外的化學藥物;另一方面也可能是減肥類保健品中經常使用的番瀉葉、蘆薈、大黃、何首烏等原料中的蒽醌類天然成分[6-8]。雖然國家規定上述含蒽醌類成分的中藥材可以作為保健食品的原料,然而其過量食用也可能對人體產生嚴重的危害[9-11],因此這些原料中的蒽醌類成分的含量亦需得到關注。此外,上述原料中的蒽醌類成分較為復雜,在實際的液相分析中時常出現蒽醌類成分與待測定的非添藥物色譜峰相重合的情況,而這很可能對疑似陽性結果的紫外光譜判斷產生一定的干擾,容易產生假陰性結果。
針對以上問題,本文擬選擇添加可能性較大的化學藥物氟西汀、比沙可啶以及番瀉葉、蘆薈、大黃、何首烏等原料的指標性成分番瀉苷A、番瀉苷B、蘆薈苷、大黃素、大黃素甲醚、大黃酸、大黃酚、蘆薈大黃素減肥類保健食品作為研究對象;利用高效液相色譜-二極管陣列檢測器(HPLC-DAD),建立一個可以同時測定上述10種化合物的定性、定量檢測方法。采用該方法對日常抽樣的減肥類保健食品進行測定,同時對疑似檢出的氟西汀和比沙可啶進行質譜確認,以驗證方法的適用性,從而為更加快捷、準確、有效的監督檢驗及更為有效的評價保健食品的質量提供技術保障。
1 材料與方法
1.1材料與儀器
日常抽樣的25種減肥類保健食品包括片劑、膠囊、減肥咖啡、袋泡茶等;番瀉苷A(94.7%)、番瀉苷B(94.9%)、蘆薈苷(95.0%)、大黃酸(100%)、氟西汀(100%)、蘆薈大黃素(97.8%)、比沙可啶(100%)、大黃素(100%)、大黃酚(99.6%)、大黃素甲醚(99.1%)標準品購自中國食品藥品檢定研究院;甲醇、乙腈均為色譜純,賽默飛世爾科技(中國)有限公司;水為超純水;其他試劑均為分析純。
Agilent1260型高效液相色譜儀配1260型分離系統、1260型二極管陣列檢測器、EZchrom軟件系統,美國安捷倫公司;Agilent6538型UPLC-DAD-ESI-Q-TOF/MS液質聯用儀美國安捷倫公司;Thermo Scientific heraeus biofuge primor離心機賽默飛世爾科技有限公司;KQ-300DA數控超聲波清洗器昆山市超聲儀器有限公司。
1.2溶液的制備
混合對照品貯備液的制備:精密稱取減壓干燥至恒重的上述標準品適量,加甲醇溶解并稀釋至刻度,搖勻,分別制得含番瀉苷A 0.288 mg/mL、番瀉苷B 0.660 mg/mL、蘆薈苷0.296 mg/mL、大黃酸0.172 mg/mL、氟西汀1.336 mg/mL、蘆薈大黃素0.246 mg/mL、比沙可啶0.572 mg/mL、大黃素0.244 mg/mL、大黃酚0.244 mg/mL、大黃素甲醚0.800 mg/mL的單標準貯備液,分別精密量取上述單標準貯備液5.0 mL混勻,即得。
供試品溶液的制備:取10倍單次用量的供試品,研細,精密稱取適量(約相當于1次用量),置于具塞三角瓶中,精密加入甲醇20 mL,稱定重量,超聲提取30 min,放冷,稱重,用甲醇補足損失的重量,搖勻,過0.45 μm濾膜,取續濾液作為供試品溶液。
陰性空白溶液的制備:稱取經測定不含有10種待測成分的供試品(秀中秀牌減肥膠囊),按照上述“供試品溶液的制備”處理,即得。
加標溶液的制備:精密稱取陰性空白樣品(秀中秀牌減肥膠囊)內容物0.5 g,置20 mL容量瓶中,分別精密加入混合對照品貯備液2.0 mL,再加入甲醇15 mL,超聲提取30 min,放冷至室溫,用甲醇定容至刻度,搖勻,靜置后取上清液過0.45 μm濾膜,平行制備6份。
1.3色譜條件
色譜柱為Diamonsil C18柱(250 mm×4.6 mm,5 μm);流動相為乙腈(A)-0.02 mol/L乙酸銨溶液(含0.4%乙酸)(B)。梯度洗脫:0~5 min,85% B;5~25 min,85% B~35% B;25~50 min,35% B;50~55 min,35% B~85% B;測定波長為268 nm;流速1.0 mL/min;柱溫25 ℃;進樣體積為10 μL。
1.4質譜條件
采用ESI源;正離子模式;霧化氣為氮氣;毛細管電壓3500 V(+);干燥氣溫度350 ℃,流速10 L/min;霧化氣壓力40 psi;碎片電壓100 eV;碰撞能量20 eV;掃描范圍m/z 100~1000。
1.5數據處理
采用液相色譜儀自帶的EZchrom軟件對實驗所得圖譜數據進行積分、命名等處理,使用Excel數據處理軟件進行統計分析。
2 結果與討論
2.1測定波長的選擇
將各對照品單標準溶液進行紫外全波長(200~400 nm)掃描,獲得了各個成分的紫外光譜圖(圖1),并將其用于樣品中疑似峰的定性判別。通過對比各成分的紫外光譜圖,最終綜合選擇紫外吸收均較強的268 nm作為定量測定波長。
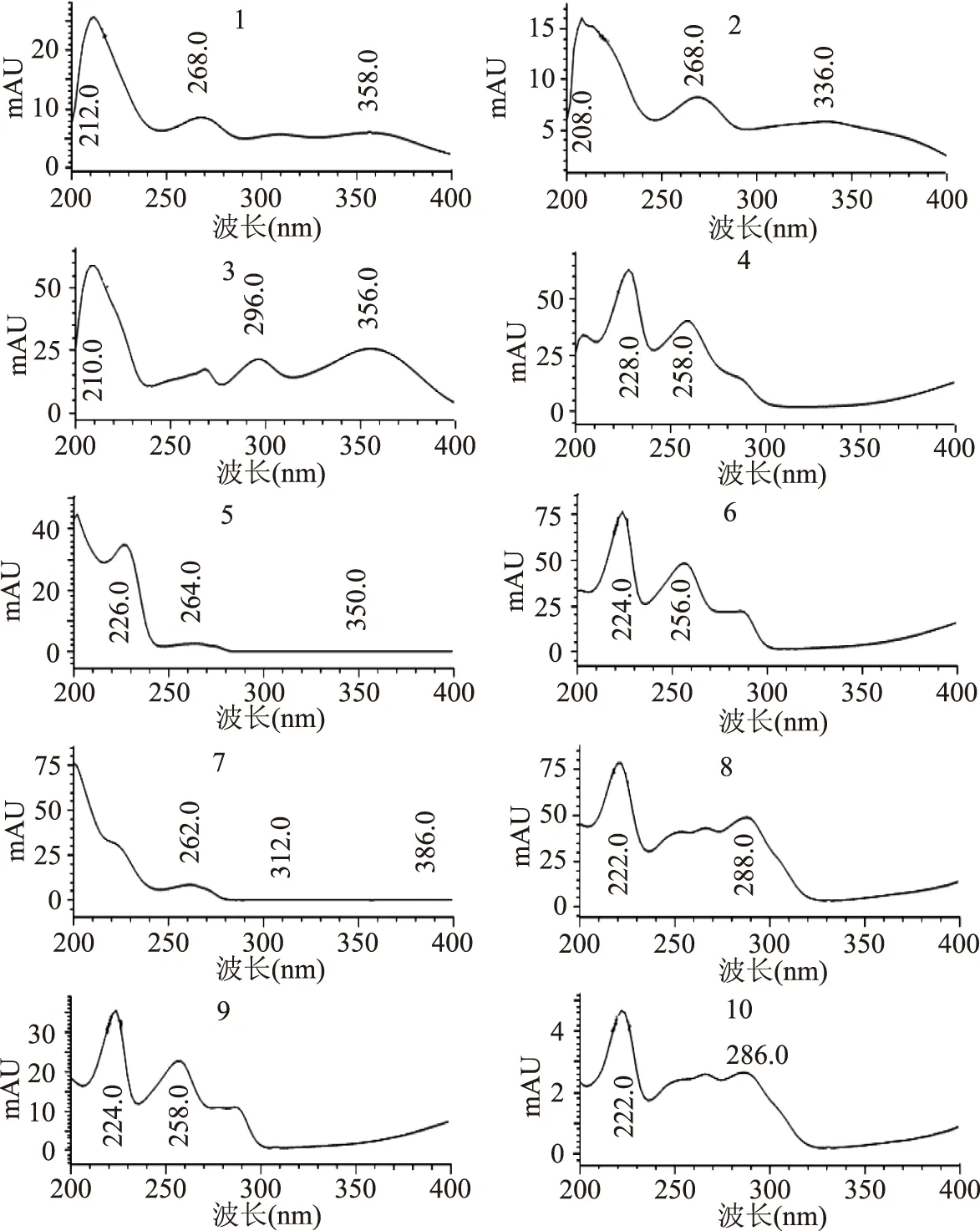
圖1 各成分紫外光譜圖Fig.1 The UV spectra of the 10 components注:1:番瀉苷B,2:番瀉苷A,3:蘆薈苷,4:大黃酸,5:氟西汀;6:蘆薈大黃素,7:比沙可啶,8:大黃素,9:大黃酚,10:大黃素甲醚。
2.2流動相的選擇
由于待分析的化合物極性相差較大,采用梯度洗脫更有利于分離。基于價格以及毒性考慮,首先采用甲醇作為有機相,然而經實驗摸索發現采用甲醇作為有機相各成分分離情況不佳,且分析時間較乙腈明顯延長,整個梯度約需80 min(見圖2)。而采用乙腈后發現各成分無論是分離效果還是峰形均較好,且分析時間明顯縮短,整個梯度約需50 min(見圖4)。因此本文采用乙腈作為有機相。
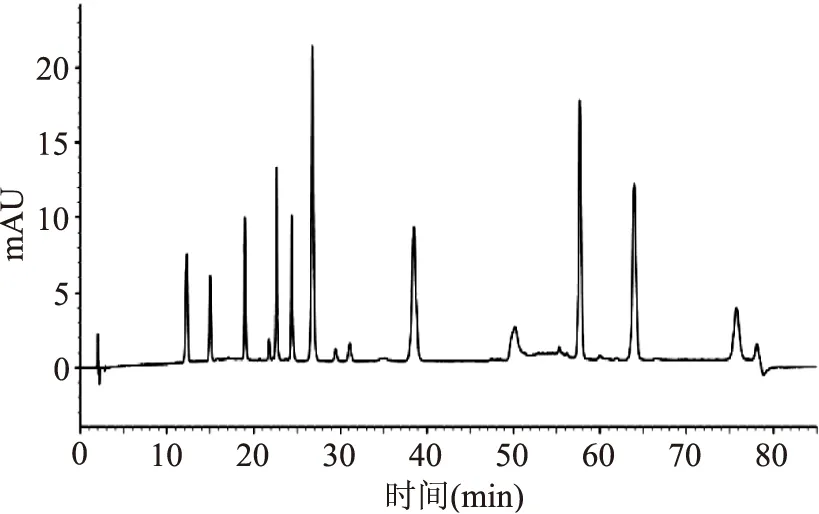
圖2 流動相為甲醇-0.02 mol/L乙酸銨溶液(含0.4%乙酸)時混標色譜圖Fig.2 The chromatogram of mixed standards for mobile phase of methanol and 0.02 mol/L ammonium acetate solution(containing 0.4% acetic acid)
為了維持各成分在色譜柱中保留的穩定性,得到較好的重現性,并提高分離選擇性及改善峰型,可以選擇向流動相中加入緩沖鹽溶液以保持體系的酸堿度。本文首先向水相中加入一定量的乙酸,結果發現峰形雖然較好,然而分離效果不佳(見圖3)。通過參考其他方法[5]并為后期質譜確認提供便捷,在上述基礎上加入了0.02 mol/L的乙酸銨,結果發現各成分得到很好地分離(見圖4)。
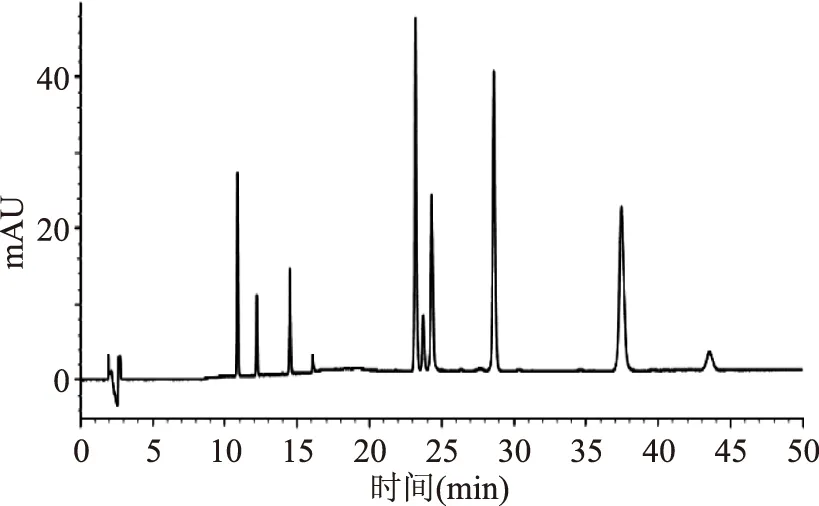
圖3 流動相為乙腈-0.4%乙酸時混標色譜圖Fig.3 The chromatogram of mixed standards for mobile phase of acetonitrile and 0.4% acetic acid solution
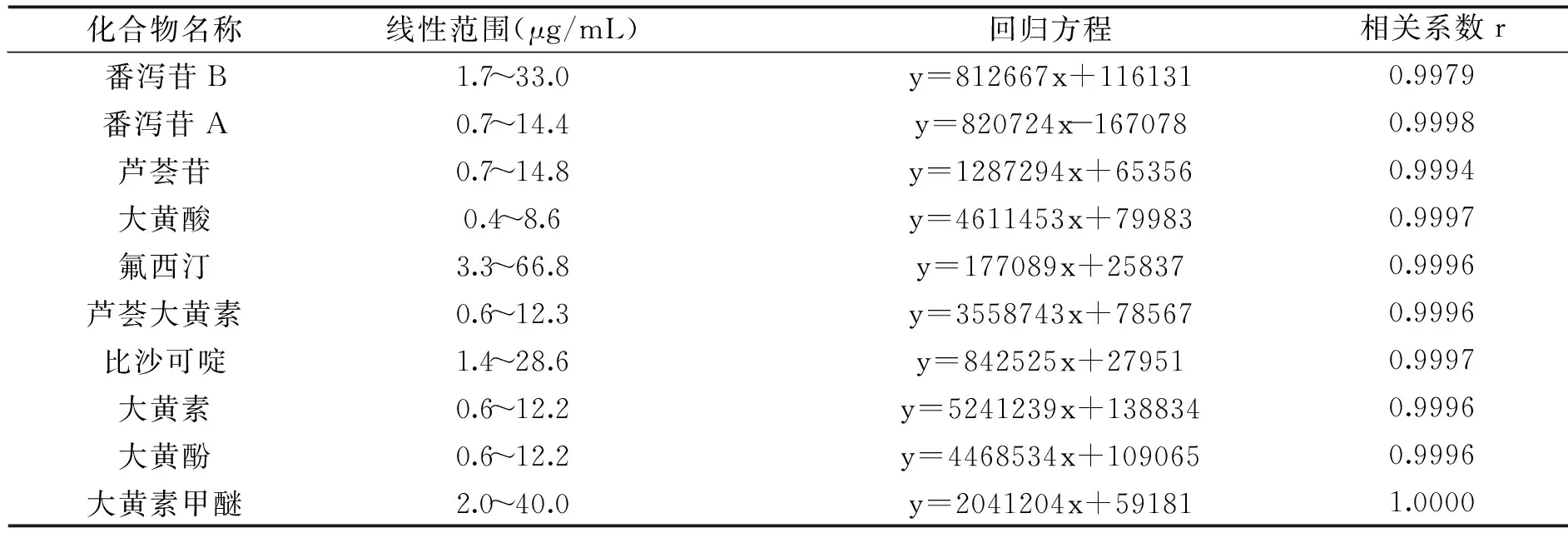
表1 10種成分的線性回歸方程
2.3方法學考察
2.3.1專屬性分別取空白提取溶劑(甲醇)、混合對照品溶液、陰性空白溶液和加標溶液進樣測定,可看出空白溶劑及陰性空白溶液對各成分的測定不產生干擾;混合對照品溶液和加標溶液在該色譜條件下10種成分均分離良好(圖4A~D)。
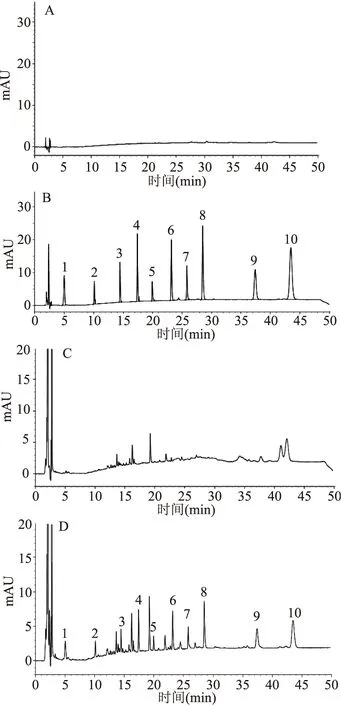
圖4 空白溶劑(A)、混合標準溶液(B)、空白供試品溶液(C)及加標溶液(D)色譜圖Fig.4 Typical chromatograms of blank(A), mixed standards solution(B),negative sample solution(C) and recovery test solution(D)注:1:番瀉苷B,2:番瀉苷A,3:蘆薈苷,4:大黃酸,5:氟西汀;6:蘆薈大黃素,7:比沙可啶,8:大黃素,9:大黃酚,10:大黃素甲醚。
2.3.2線性關系考察精密量取1.2項“混合對照品貯備液”0.5、1.0、2.0、5.0和10 mL,分別置于20 mL容量瓶中,加甲醇稀釋至刻度,搖勻,作為標準曲線溶液。分別吸取10 μL進樣測定,記錄色譜圖,以各成分峰面積(Y)對濃度(X,μg/mL)進行線性回歸,回歸方程、相關系數見表1。結果發現相關系數均大于0.995,表明各成分的線性關系較好。
2.3.3檢測限、定量限將混合對照品貯備液進行一系列的稀釋后測定,在信噪比S/N約為3時測得檢測限,在信噪比S/N約為10時測得定量限,各成分的檢出限、定量限結果見表2。結果表明,各成分檢出限均低于1 μg/mL,定量限均低于2 μg/mL,方法的靈敏度可以達到含量測定的要求。
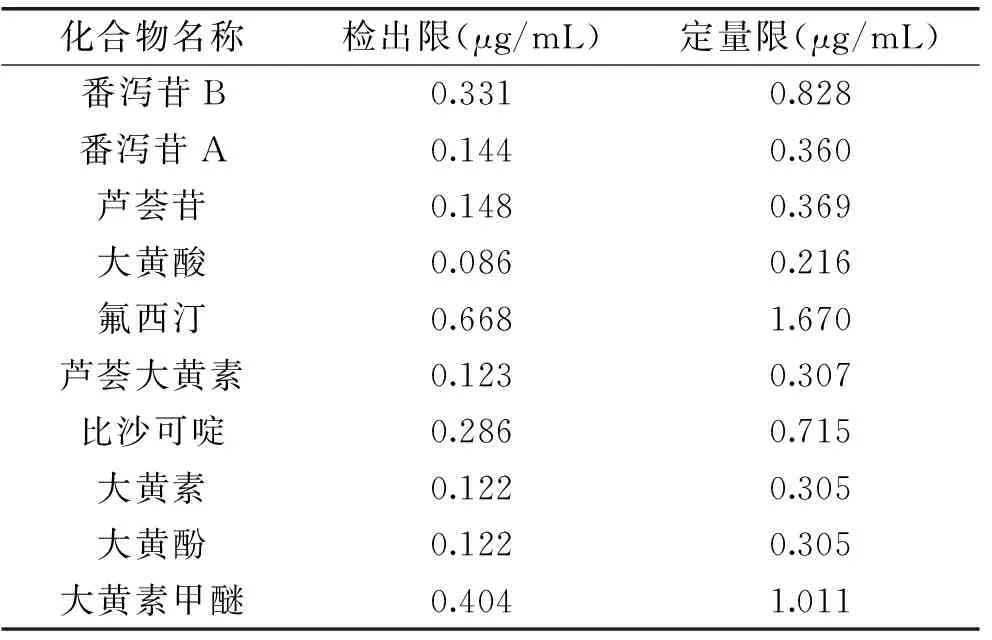
表2 10種成分的檢出限、定量限結果
2.3.4精密度精密量取混合對照品貯備液1.0 mL置20 mL容量瓶中,加甲醇稀釋至刻度,搖勻,重復進樣6次,記錄色譜圖,計算各個成分峰面積的相對標準偏差,結果發現各個成分的RSD均小于2%,方法精密度較好。
2.3.5對照品溶液穩定性取2.3.4項精密度實驗溶液,分別于配制后的第0、1、2、4、12、24 h進樣測定,記錄色譜圖并計算各個成分峰面積的RSD。結果表明10種成分24 h內峰面積的RSD均不大于3%,表明對照品溶液穩定性良好。
2.3.6準確度采用加標回收實驗考察方法的準確度。取“1.2項”制備的加標溶液進樣測定,計算各個成分的回收率(見表3)。結果發現10個成分回收率均位于90.3%~104.7%之間,RSD均小于5.0%,表明方法能準確的對各成分進行定量測定。
2.3.7供試品溶液穩定性取加標回收溶液,分別于配制后的第0、1、2、4、12、24 h進樣測定,記錄色譜圖并計算各成分峰面積的RSD。結果發現10種成分峰面積的RSD均小于3%,表明供試品溶液中各成分穩定性良好。
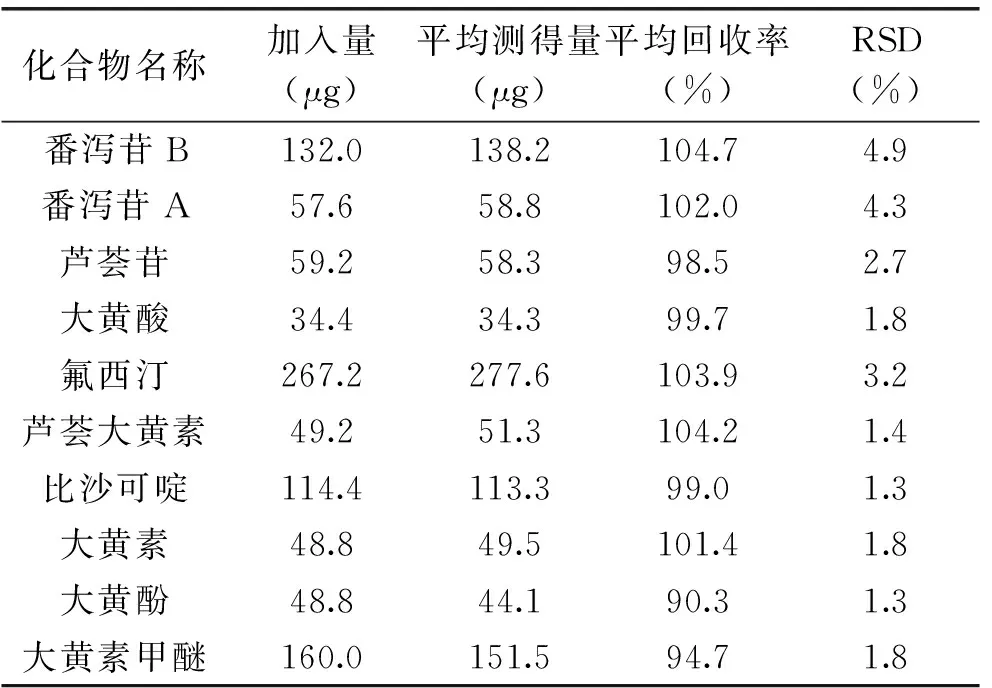
表3 加標回收率實驗結果(n=6)
2.3.8耐用性本文考察了不同柱溫、不同流速及不同廠家色譜柱對分離的影響程度。將原色譜條件中的柱溫由25 ℃分別調整為20 ℃和30 ℃,流速由1.0 mL/min調整為1.1 mL/min和0.9 mL/min,分別取混合對照品溶液進樣,結果發現10種成分在相應條件下均能得到很好地分離(見圖5A~D)。
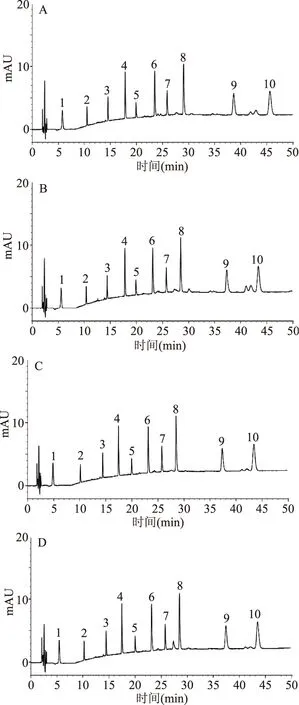
圖5 耐用性實驗下混標溶液色譜圖Fig.5 The chromatograms of mixed standards for durability tests注:A柱溫20 ℃,B柱溫30 ℃;C流速1.1 mL/min,D流速0.9 mL/min。
分別取混和對照品溶液,按“1.3項”色譜條件使用Inertsil ODS-3型C18柱(4.6 mm×250 mm,5 μm)進行測定,結果發現對照品溶液各成分在該色譜柱也可以達到基線分離(見圖6)。結果表明本方法在不同柱溫、流速及不同廠家色譜柱上10種成分均能夠得到良好地分離,表明方法的耐用性較好。
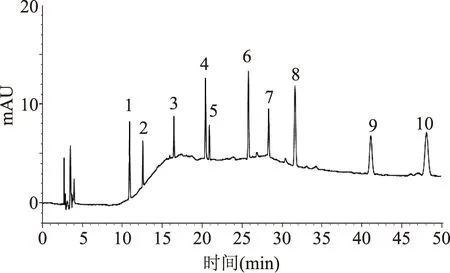
圖6 Inertsil ODS-3型色譜柱上混標溶液色譜圖Fig.6 The chromatogram of mixed standards using Inertsil ODS-3 column
2.4樣品測定
取25種日常抽檢的減肥類保健食品,按1.2項“供試品溶液的制備”方法進行處理并測定(若測得成分濃度不在線性范圍內則將供試品溶液進行稀釋),以保留時間和紫外光譜圖對檢出峰進行初篩,并對疑似檢出的非添成分(比沙可啶、氟西汀)進行一級掃描及二級碎片離子(質譜圖見圖7、圖8)比對確認。
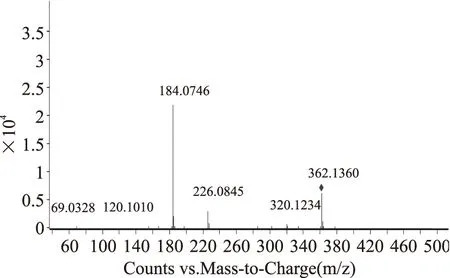
圖7 比沙可啶對照品準分子離子的二級質譜圖Fig.7 ESI-MS2 product ion spectra of bisacodyl
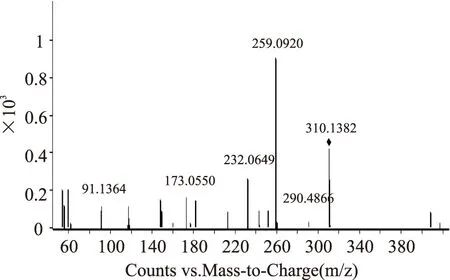
圖8 氟西汀對照品準分子離子的二級質譜圖Fig.8 ESI-MS2 product ion spectra of fluoxetine
從表4發現2種樣品中檢出氟西汀,1種樣品中檢出比沙可啶。氟西汀和比沙可啶并不在當前國標檢測的范圍內,由此提示對國家標準進行修訂的必要性。1種樣品中同時檢出蘆薈苷和蘆薈大黃素(該樣品標簽中標示含有蘆薈),二者含量分別為16120 mg/kg和120 mg/kg。
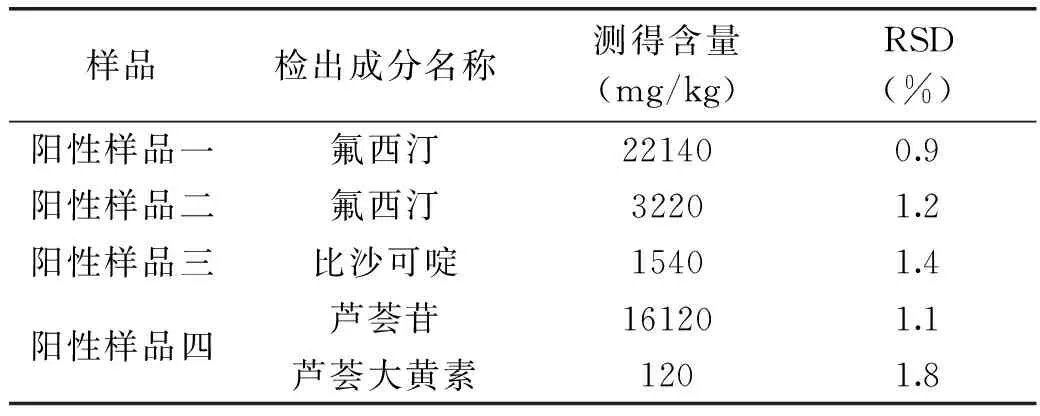
表4 樣品中非法添加藥物及部分蒽醌類成分含量測定結果(n=3)
3 結論
本文建立了一種可以同時測定減肥類保健食品中可能添加的化學藥物氟西汀、比沙可啶以及8種蒽醌類中藥成分的分析方法。方法學考察發現10種待測物檢出限均低于1 μg/mL;線性關系良好(r≥0.995);平均回收率位于90.3%~104.7%之間,RSD(n=6)均小于5.0%;各成分在不同柱溫、不同流速及不同廠家色譜柱上均達到了基線分離。上述方法學驗證結果表明方法分離效果好,具有較高的選擇性和靈敏度,耐用性較強。在對日常抽樣的25種減肥類保健食品檢測發現,有2種樣品檢出氟西汀,1種樣品檢出比沙可啶,另有1種樣品中同時檢出中藥成分蘆薈苷和蘆薈大黃素,由此表明方法的針對性較強,適用性良好,可以用于減肥類保健食品中非添藥物的測定及蒽醌類中藥成分的定性、定量分析。
[1]孫鑫貴,趙榕,張正,等.2005-2008年全國減肥類保健食品違法添加藥物狀況調查及分析[J].中國食品衛生雜志,2010,22(5):453-455.
[2]王靜文,黃湘鷺,曹進,等.超高效液相色譜法同時測定減肥類保健食品中非法添加的25種藥物[J].色譜,2014,32(2):151-156.
[3]國家食品藥品監督管理局.藥品檢驗補充檢驗方法和檢驗項目批準件2006004[S].2006.
[4]國家食品藥品監督管理局.食藥監辦許[2010]114號文件附件2:減肥類保健食品違法添加藥物的檢測方法[S].2010.
[5]國家食品藥品監督管理局.藥品檢驗補充檢驗方法和檢驗項目批準件2012005[S].2012.
[6]肖晶,楊杰,高尚偉,等.HPLC法測定保健食品中蒽醌類成分的含量[J].中國食品衛生雜志,2010,22(1):27-30.
[7]汪鳳云,李紅艷,鞏飚.HPLC法測定蘆薈保健制品中蘆薈苷的含量[J].中國衛生檢驗雜志,2008,18(11):2238-2239.
[8]莫紫梅,朱斌.高效液相色譜法測定保健食品中番瀉苷含量的研究[J].中國衛生檢驗雜志,2014,24(5):2238-2239.
[9]余翠琴,張凡.慎用瀉藥[J].中國臨床藥學雜志,2002,11(5):307-308.
[10]李軍生,鄒義英.蘆薈類保健食品的毒理學安全性評價[J].毒理學雜志,2009,23(3):253-254.
[11]李衛東.長期應用番瀉葉對大鼠結腸電及Cajal間質細胞的影響[J].廣州中醫藥大學學報,2005,22(5):408-409.
Determination of rhubarb anthraquinones and illegally added chemical drugs in weight-loss functional foods
LU Hui,QIAN Ye-fei,ZHANG Bin,HAO Gang,CHEN Li-bo*
(Suzhou Institute for Food and Drug Control,Suzhou 215104,China)
An analytical method using high performance liquid chromatography coupled with diode array detector(HPLC-DAD)was developed for determination of illegally added chemical drugs fluoxetine and bisacodyl,and rhubarb anthraquinones such as sennoside A,sennoside B,emodin,emodin monomethyl ether,rheinic acid,chrysophanol,barbaloin and aloe-emodin in weight-loss functional foods. The separation was performed on a Diamonsil C18column(250 mm×4.6 mm,5 μm)by gradient elution of acetonitril and 0.02 mol/L ammonium acetate solution(containing 0.4% acetic acid)at a flow rate of 1.0 mL/min:0~5 min,85% B,5~25 min,85% B~35% B,25~50 min,35% B,50~55 min,35% B~85% B. The detection wavelength was 268 nm and the column temperature was 25 ℃. The samples containing fluoxetine or bisacodyl were confirmed by two grade of fragmention comparison using electrospray ionization-quadrupole-time of flight mass spectrometer(ESI-Q-TOF/MS). The correlation coefficient of standard curve for each component in linearity range was not less than 0.995,as well as the average recoveries were 90.3%~104.7% with the relative standard deviations(RSDs)were not more than 5.0%. After determination of 25 weight-loss functional foods,the fluoxetine and bisacodyl were detected in 2 and 1 samples,respectively. The barbaloin and aloe-emodin were both detected in one of the 25 samples. The method is reliable,sensitive,accurate and reproducible. It can be used for the qualitative and quantitative determination of above 10 components in weight-loss functional foods.
HPLC-DAD;weight-loss functional foods;illegally added;fluoxetine;bisacodyl;anthraquinones
2015-10-26
魯輝(1982-),男,碩士,主管藥師,研究方向:保健食品、化妝品檢測與質量控制,E-mail:15862509876@163.com。
陳麗波(1969-),女,主任藥師,主要從事保健食品、化妝品質量控制與評價方面的研究,E-mail:chenlb@szifdc.org.cn。
蘇州市科技計劃項目-應用基礎研究(SYS201583)。
TS207.3
A
1002-0306(2016)10-0088-06
10.13386/j.issn1002-0306.2016.10.008
猜你喜歡
中學生百科·大語文(2023年2期)2023-05-08 14:11:47
故事作文·低年級(2023年1期)2023-02-23 07:19:20
小讀者(2021年6期)2021-07-22 01:50:02
科教新報(2021年23期)2021-07-21 14:37:25
故事作文·低年級(2021年4期)2021-05-06 03:11:13
作文評點報·小學三、四年級(2019年14期)2019-04-30 00:22:22
故事作文·低年級(2016年1期)2016-09-10 07:22:44
故事作文·高年級(2015年5期)2015-09-08 08:27:03
江蘇調味副食品(2015年1期)2015-02-28 01:56:34
故事作文·低年級(2009年5期)2009-05-06 03:35:50
