離子色譜-柱后衍生-紫外可見檢測法測定地下水中痕量六價鉻
2016-12-30 01:23:12周健楠楊懂艷丁萌萌劉保獻
分析儀器 2016年6期
關鍵詞:方法
徐 碩 周健楠 楊懂艷 丁萌萌 常 淼 劉保獻
(北京市環境保護監測中心,北京 100048)
?
離子色譜-柱后衍生-紫外可見檢測法測定地下水中痕量六價鉻
徐 碩1周健楠 楊懂艷 丁萌萌 常 淼 劉保獻
(北京市環境保護監測中心,北京 100048)
建立了離子色譜-柱后衍生-紫外可見檢測法直接測定地下水中痕量六價鉻的方法,確定了方法的性能指標,開展了干擾實驗的研究。研究發現,本方法可在8分鐘內完成分析,六價鉻峰型尖銳,方法標準曲線線性范圍0.05~5.00 μg/L,相關系數≥0.9999。方法檢出限較低(0.008 μg/L),精密度和準確度較高,3種地下水實際樣品的加標回收率為94%~106%,RSD為0.6%~4.0%(n=8)。高濃度SO42-(500~3000 mg/L)對六價鉻測定無干擾,而Cl-(>500 mg/L)可在一定程度上干擾六價鉻峰形。本方法耗時短、選擇性強、靈敏度高、前處理簡單、干擾較少,相對常規方法能夠更好地滿足地下水中六價鉻測定的實際需要。
離子色譜 柱后衍生 地下水 六價鉻
六價鉻Cr(Ⅵ)在天然水體中具有很好的溶解性,多以CrO42-和HCrO4-的形式存在。六價鉻對人類、動物及水生生物均有毒害作用,其半致死量ED50是三價鉻的100倍,被國際癌癥研究署(IARC)列為一級致癌物[1]。我國《地下水質量標準》(GB/T 14848-93)中明確規定六價鉻的I類限值為0.005 mg/L[2]。
目前,地下水中六價鉻的測定方法主要有二苯碳酰二肼分光光度法、離子色譜法、流動注射分光光度法、原子吸收光譜法、原子熒光光譜法以及ICP-AES等,其中國家標準(GB 7467-87)[3]采用二苯碳酰二肼分光光度法,最低檢出濃度0.004 mg/L,雖略低于I類限值,但無法滿足地下水中痕量六價鉻的定量檢測,且該方法易受樣品濁度、色度及干擾離子的影響,容易出現假陽性或假陰性結果、重現性差。
離子色譜法可有效排除樣品基體干擾,色譜柱的分離作用使得六價鉻單獨進入檢測器。與常見的電導檢測器相比,柱后衍生紫外可見檢測可以特異性地檢出六價鉻,而對其他陰離子無響應,因此在測定時干擾更少、靈敏度更高。本實驗采用離子色譜法直接測定地下水中的六價鉻,水樣中以鉻酸鹽(CrO42-)形式存在的六價鉻經陰離子色譜柱與樣品基體分離,由在線柱后衍生,流入紫外可見檢測器,于530 nm處檢測,考察了方法的準確性、重現性和實用性,獲得了方法的性能指標。
1 實驗部分
1.1 試劑與儀器
Dionex ICS-3000型離子色譜儀(美國賽默飛世爾科技公司),配有柱后衍生裝置、Dionex Ultimate 3000可變波長紫外可見檢測器;Chromeleon 6.8色譜工作站; 0.45μm微孔濾膜過濾頭(水系)。
硫酸銨(分析純,國藥集團化學試劑有限公司);氨水(優級純,國藥集團化學試劑有限公司);二苯碳酰二肼(分析純,國藥集團化學試劑有限公司);甲醇(HPLC級,TEDIA);濃硫酸(優級純,北京化工廠);六價鉻標準溶液(100μg/mL,國家標準物質研究中心);實驗用水均為新制備的二次去離子水,電阻率>18.2 MΩ·cm。
1.2 色譜條件
色譜柱:IonPac AS7分離柱(4 mm×250 mm),IonPac NG1保護柱(4 mm×35 mm);淋洗液:250 mmol/L 硫酸銨-100 mmol/L 氨水,1.0 mL/min;柱后衍生劑:2 mmol/L 二苯碳酰二肼(DPC)-0.5 mol/L 硫酸-10%(V/V)甲醇,0.30 mL/min;進樣體積:900 μL;檢測波長:530 nm。
1.3 標準曲線繪制
取六價鉻標準溶液,配制成0.05μg/L、0.20 μg/L、0.50 μg/L、1.00 μg/L、2.00 μg/L、5.00μg/L共6個濃度的標準系列,按上述色譜條件進行測定。以六價鉻濃度為橫坐標,儀器響應值(峰面積)為縱坐標繪制標準曲線。
1.4 樣品制備
地下水樣品采集后于0~4℃運輸或保存,24 h內測定,測定前使用0.45 μm濾膜過濾,舍去1~2mL初濾液,收集續濾液,置于自動進樣器樣品瓶中,待測。
2 結果與討論
2.1 六價鉻標準譜圖
依照1.2節所述色譜條件,測定1.0μg/L的六價鉻標準溶液,標準譜圖如圖1所示。方法可在8 min內完成分析,六價鉻保留時間在5.9 min附近,峰形尖銳,周圍無雜峰,方法的色譜條件可滿足六價鉻的檢測需求。
2.2 方法線性及檢出限
根據1.3所述標準曲線繪制方法,開展方法標準曲線線性實驗,結果表明六價鉻質量濃度在0.05~5.00μg/L時,峰面積Y與六價鉻濃度X(μg/L)線性關系良好,線性回歸方程為Y=0.347X,相關系數r=0.9999。標準曲線如圖2所示。
根據《環境監測分析方法標準制修訂技術導致》(HJ168-2010)[4],配制濃度為0.05 μg/L的六價鉻標準溶液,按上述色譜條件平行測定7次,結果見表1。經計算,本方法直接測定六價鉻的方法檢出限為0.008 μg/L,測定下限為0.032 μg/L,比國標方法GB 7467-87的最低檢出濃度(0.004 mg/L)低兩個數量級,可很好地滿足《地下水質量標準》(GB/T 14848-93)[2]中六價鉻的測定需求。

表1 方法檢出限測定結果
2.3 精密度和準確度
為考察實驗室內空白加標樣品的精密度和準確度,使用純水分別配制0.04μg/L、1.00 μg/L、5.00 μg/L的六價鉻標準溶液,平行測定6次,RSD值在0.7%~3.7%之間(見表2);同時將濃度為500 μg/L和54.9 μg/L的有證標準物質稀釋100倍,平行測定6次,RE值分別為0.2%和2.6%(見表3)。
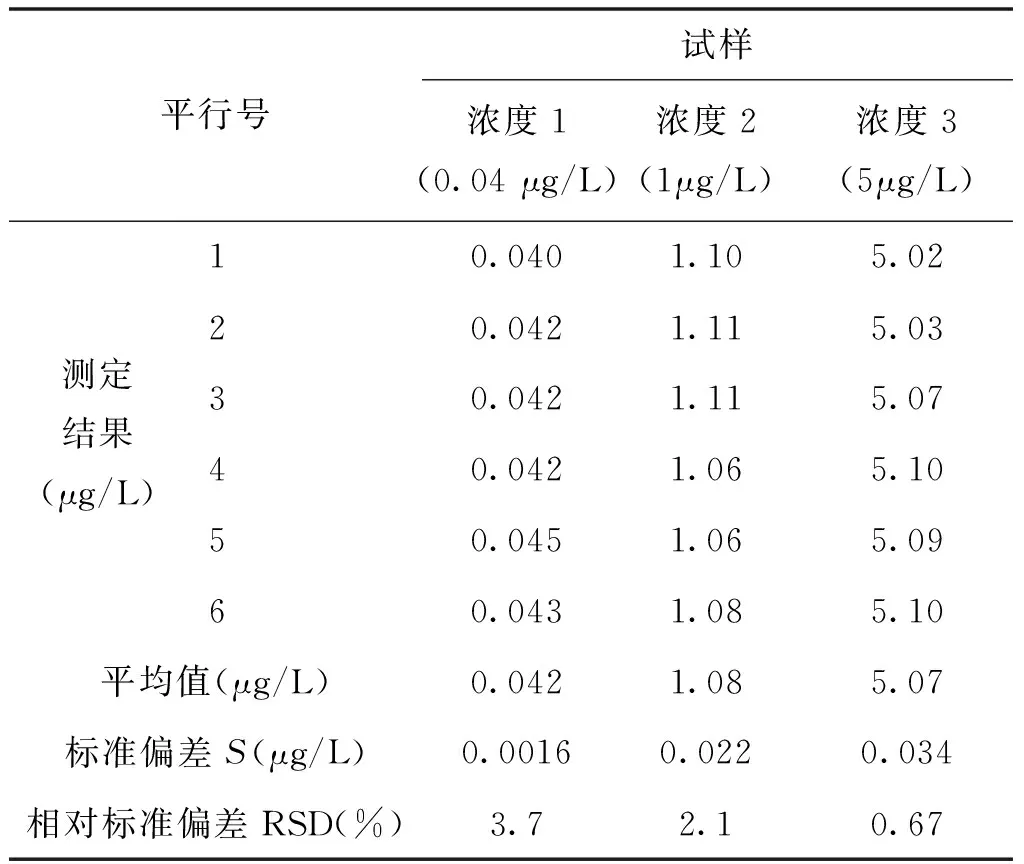
表2 方法精密度測定結果
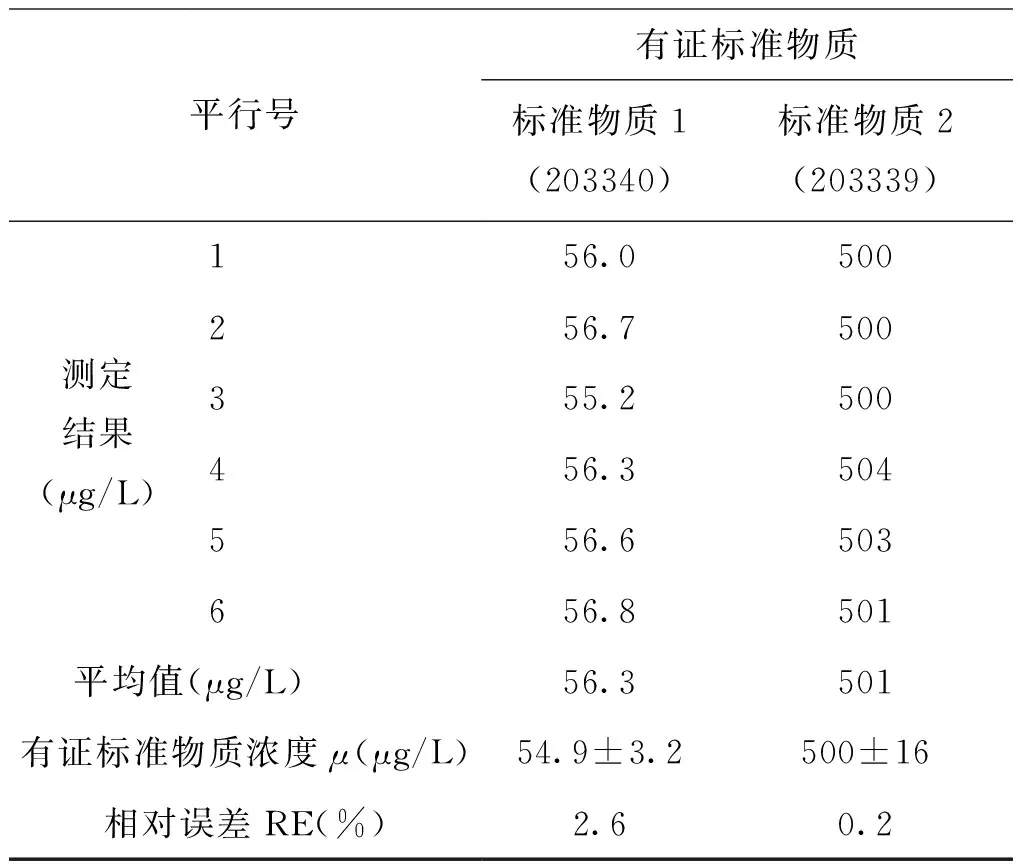
表3 方法準確度測定結果
為考察本方法測定不同種類實際地下水的精密度和準確度,選取3種地下水井(平原區域地下水井、工業區水井、水源井)采集水樣。樣品連續進樣測定8次,RSD為0.6%~4.0%,樣品加標后測得回收率為94%~106%(見表4)。由此可見,采用離子色譜-柱后衍生-紫外可見檢測法直接測定地下水中痕量六價鉻,方法精密度和準確度較高,可滿足實際檢測需求。

表4 實際樣品測定結果及其加標回收率
2.4 方法干擾試驗
根據《水質六價鉻的測定二苯碳酰二肼分光光度法》(GB 7467-87)[3],濁度、色度及干擾離子(鐵、鉬、汞、釩等)都會影響六價鉻的顯色。本實驗法利用離子色譜柱將六價鉻由樣品基體中分離出來,有效排除了上述因素的干擾。
高濃度陰離子(如Cl-、SO42-)可導致色譜柱過載,表現為加標回收率降低和色譜峰拖尾。實驗室內對北京市地下水樣品的測定表明Cl-、SO42-的濃度一般為0~300 mg/L。查閱資料可知普通地下水Cl-、SO42-的濃度一般在500 mg/L以下[5,6];但部分污灌區地下水、礦區地下水、鹽化地下水中Cl-、SO42-濃度接近3000mg/L[7]。不同濃度陰離子(Cl-、SO42-)對六價鉻(1.00 μg/L)測定的影響如圖3所示。可見,隨著Cl-濃度的增加,六價鉻峰高逐漸降低,但峰面積不變,即出現峰形變寬變矮的現象。SO42-濃度的增加并未對六價鉻峰形造成顯著影響,峰面積和峰高變化不大。因此,地下水中六價鉻的測定可能會受到高濃度Cl-(>500 mg/L)的干擾,實際測定時應考察六價鉻峰形及加標回收率。
3 結論
地下水中六價鉻濃度一般為10-9(μg/L)級別甚至更低。在地下水的實際測定中,目前普遍采用的二苯碳酰二肼分光光度法對大部分水樣的測定結果均是未檢出。本實驗所用的離子色譜-柱后衍生-紫外可見檢測法,檢出限低至0.008 μg/L,可檢測出地下水中六價鉻的實際濃度,為相關的痕量六價鉻研究提供了可靠的手段。在已配備有離子色譜儀的實驗室中,此方法可快速建立,簡單易行。對于以水質評判為目的的六價鉻監測,離子色譜-柱后衍生-紫外可見檢測法相對于二苯碳酰二肼分光光度法的優勢在于能夠連續自動進樣分析,且不受樣品基體干擾,十分適用于大量水樣或干擾物質較多的不清潔水樣的定量檢測。但對于少量的清潔水樣,由于離子色譜法試劑需現用現配,采用二苯碳酰二肼法更為合適。
水中六價鉻的其它測定方法中,流動注射分光光度法需要復雜的預處理來排除濁度、色度等樣品基體的干擾[8-10];原子吸收光譜法、原子熒光光譜法、電感耦合等離子光譜及質譜法只能測定總鉻,或需要繁瑣的離子分離步驟才能測定六價鉻,影響分析速度[11];離子色譜分離-電導檢測法的檢出限為5 μg/L[12],改進后的毛細管離子色譜分離-電導檢測法檢出限低至1.0 ng/L,但分析時間長達41 min,同時其樣品色譜圖中雜峰較多,六價鉻響應信號并不突出[13]。
本實驗選用的離子色譜-柱后衍生-紫外可見檢測法,可將樣品過濾后直接進樣,分析耗時8 min;六價鉻峰形尖銳,周圍無雜峰;經標準樣品及實際樣品測定,方法具有非常好的準確性及重現性。因此,本方法在水中痕量六價鉻的檢測方面具有良好的應用前景。
[1] IARC.IARC monographs on the evaluation of carcino-genic risks to humans volume 49 chromium, nickel and welding[R].Geneva:World Health Organization,1997:17-33.
[2] GB/T 14848-93,地下水質量標準[S].
[3] GB 7467-87,水質六價鉻的測定二苯碳酰二肼分光光度法[S].
[4] HJ 168-2010,環境監測分析方法標準制修訂技術導則[S].
[5] 劉春,譚利敏,尹國勛,范俊玲.焦作市某污灌區地下水無機氯化物污染原因初探[J].江蘇環境科技,2006,19(z2):124-126.
[6] 倪傳鈞.淮南潘集礦區地下水水化學特征分析[J].安徽科技,2006(2):44-46.
[7] 李彬,史海濱,張建國,李禎.節水改造前后內蒙古河套灌區地下水水化學特征[J].農業工程學報,2014,30(21):99-110.
[8] ISO23913:2006,Water quality — Determination of chromium(Ⅵ)— Method using flow analysis (FIA and CFA)and spectrometric detection [S].
[9] 林志鵬,劉海術,楊芳.流動注射分光光度法測定水中六價鉻的研究[J].干旱環境監測,2014,28(2):66-69, 93.
[10] 樊靜,陳亞紅,馮素玲.流動注射在線分離預濃集分光光度法測定環境水樣中的痕量鉻(Ⅵ)[J].分析試驗室,2004,23(1):70-72.
[11] 丁紅紅,鄭明凱.飲用水中六價鉻測定方法進展[J].廣州化工,2012,40(17):39-40, 82.
[12] 嚴利民,胡文武.離子色譜法測定水中六價鉻[J].中國熱帶醫學,2007,7(1):87-88.
[13] 胡忠陽,汪瓊,葉明立,梁立娜,何世偉,朱巖.離子色譜法測定飲用水中的六價鉻[J].中國無機分析化學,2012,02(z1):1-2.
Determination of trace Cr(Ⅵ) in groundwater by ion chromatography(IC) with post-column derivatization and UV/Visible spectrometer.
Xu Shuo, Zhou Jiannan, Yang Dongyan, Ding Mengmeng, Chang Miao,Liu Baoxian
(BeijingMunicipalEnvironmentalMonitoringCenter,Beijing100048,China)
The method was proved to be effective in Cr(Ⅵ) determination, and the analysis was accomplished in 8 minutes with a favorable peak profile. The correlation coefficient was >0.9999 for Cr(Ⅵ) in the range of 0.05-5 μg/L, and the detection limit was as low as 0.008 μg/L. The high accuracy and precision were proved by the satisfactory recovery rates (94%-106%) and RSDs (0.6%-4.0%) for 3 different kinds of actual groundwater samples. The high concentration of sulfate had no influence in Cr(Ⅵ) detection while the high concentration of chloride could compromise the chromatography.This method can be used for Cr(Ⅵ) determination in groundwater.
ion chromatography;post-column derivatization;groundwater;Cr(Ⅵ)
徐碩,女,1990年出生,工程師,碩士研究生,從事環境監測工作,E-mail:shuoxu12@163.com。
劉保獻,男,1983年出生,高級工程師,碩士研究生,從事環境監測工作,E-mail:liubaoxian28@163.com。
10.3936/j.issn.1001-232x.2016.06.008
2016-03-14
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
