依普利酮部分逆轉甲狀腺激素誘導的心肌電重構
2017-04-11 04:38:43易甜甜羅水蓮
中國病理生理雜志 2017年3期
趙 璐, 易甜甜, 羅水蓮, 蘇 立
(重慶醫科大學附屬第二醫院心內科,重慶 400010)
依普利酮部分逆轉甲狀腺激素誘導的心肌電重構
趙 璐, 易甜甜, 羅水蓮, 蘇 立△
(重慶醫科大學附屬第二醫院心內科,重慶 400010)
目的: 研究醛固酮拮抗劑依普利酮對甲狀腺激素(T3)誘導的心肌電重構的影響。方法:原代培養SD乳鼠心肌細胞,并隨機分為對照組、T3組、依普利酮(Epl)組、T3+Epl組,采用免疫熒光法鑒定心肌細胞,CCK-8法檢測T3與依普利酮對心肌細胞存活率的影響,細胞免疫熒光法、real-time PCR法、Western blot法檢測各組鉀離子通道Kv1.5、Kv4.3,L型鈣離子通道Cav1.2,縫隙連接蛋白40(Cx40)和Cx43 mRNA與蛋白的表達。結果:免疫熒光法顯示所培養細胞為心肌細胞且95%以上橫紋肌α-輔肌動蛋白(sarcomeric α-actinin, α-actinin)抗體染色陽性。與對照組比較,T3增加Kv1.5、Kv4.3、Cav1.2與Cx40 mRNA與蛋白的表達,減少Cx43 mRNA與蛋白的表達;Epl組Kv1.5、Kv4.3、Cav1.2與Cx40 mRNA與蛋白的表達降低,Cx43的表達升高;與T3組比較,T3+Epl組Kv1.5、Kv4.3、Cav1.2與Cx40 mRNA與蛋白的表達降低,Cx43的表達升高。結論:依普利酮可部分逆轉甲狀腺激素導致的心肌電重構。
心肌細胞; 甲狀腺激素; 依普利酮; 離子通道; 縫隙連接蛋白
心肌電生理特性取決于諸多離子通道的離子交換功能,離子通道是心肌細胞電活動產生的基礎[1]。研究證實L型鈣電流(L-type Ca2+current,ICa-L)、瞬時外向鉀電流(transient outward K+current,Ito)、超快延遲整流鉀電流(ultrarapid delayed rectifier K+current,IKur)與心肌電活動密切相關。同時,心肌細胞間的電興奮通過縫隙連接進行傳導,而縫隙連接蛋白40 (connexin 40, Cx40)與Cx43是細胞間縫隙連接的主要蛋白。因此,心肌細胞離子通道與縫隙連接的改變均可導致心肌的電重構。
甲狀腺激素(thyroid hormone, T3)等體液因素可導致心肌細胞的電重構。動物實驗發現L-甲狀腺素可使大鼠心肌肥大,肥大的心肌質膜的表面積增大,離子通道相對密度減少的同時也導致多個離子通道蛋白的表達異常,出現強的致心律失常性[2]。近年來,腎素-血管緊張素-醛固酮系統(renin-angiotensin-aldosterone system,RAAS)參與心肌重構得以證實[3]。依普利酮(eplerenone,Epl)作為新型選擇性醛固酮受體拮抗劑,與鹽皮質激素受體結合,抑制醛固酮受體復合物的形成,從而影響由醛固酮受體復合物引起的一系列病理反應,且與性激素相關的副作用較螺內酯少,因此受到廣泛關注[4]。但依普利酮是否能逆轉甲狀腺激素誘導的心肌電重構尚無明確定論。因此,本研究擬通過甲狀腺激素處理原代心肌細胞,觀察依普利酮對甲狀腺素所致心肌電重構的影響。
材 料 和 方 法
1 材料
1.1 實驗動物 SPF級新生1~3 d齡SD乳鼠,雌雄不拘,由重慶醫科大學實驗動物中心提供,動物合格證編號為CQLA-2014-0187。
1.2 主要藥物與試劑 高糖DMEM培養基、胎牛血清(Gibco);Ⅱ型膠原酶、5-溴脫氧尿苷(5-bromodeoxyuridine,5-BrdU)、甲狀腺激素、胰蛋白酶(Sigma-Aldrich);依普利酮(Aladdin);CCK-8試劑盒(Dojindo);抗橫紋肌α-輔肌動蛋白(α-actinin)、Kv1.5抗體(Santa Cruz);抗Kv4.3、Cav1.2抗體(Novus);抗Cx40、Cx43 抗體(BIOSS);抗GAPDH 抗體(天德悅);Cy3標記的山羊抗兔抗體、FITC 標記的山羊抗兔抗體、辣根過氧化物酶HRP標記的抗體、DAPI染色液(碧云天);TRIzol試劑盒、逆轉錄試劑盒、real-time PCR 試劑盒(TaKaRa)。
2 方法
2.1 原代心肌細胞的分離培養與鑒定 取1~3 d齡的SD乳鼠,在無菌條件下取下乳鼠心肌組織,用0.125%胰蛋白酶及0.08% Ⅱ型膠原酶多次混合消化后離心,收集細胞懸液,接種于6孔板中,采用差速貼壁法和5-BrdU純化心肌細胞,用含有10%胎牛血清的高糖DMEM培養基,于37 ℃、5% CO2培養箱培養,40 h后換液。采用細胞免疫熒光法鑒定原代心肌細胞。
2.2 CCK-8法檢測T3與依普利酮對心肌細胞活力的影響 按每孔接種1×104個細胞的密度將心肌細胞接種于96孔板。分別加入含0、1、2、5、10、100 nmol/L T3的培養基和含0、10-8、10-7、10-6、10-5mol/L依普利酮的培養基,分別在培養24 h后加入10 μL CCK-8溶液,繼續培養2 h,使用酶標儀于450 nm處讀取吸光度(A)值。細胞存活率=(A實驗組-A空白孔)/(A對照組-A空白孔)×100%。每組設5個復孔,空白孔為未加細胞懸液的培養基。根據此結果選取最適T3和依普利酮的處理濃度。
2.3 實驗分組 實驗以原代培養3 d后的心肌細胞作為研究對象。實驗前更換無血清培養基處理24 h,使細胞生長趨于同步化,再加入各干預因素培養24 h。實驗分為4組,分別為對照(control)組、T3(10 nmol/L)組、Epl(10-6mol/L)組和T3+Epl組(先加入10 nmol/L T3,30 min后加入10-6mol/L依普利酮)。
2.4 細胞免疫熒光檢測 將無菌蓋玻片置于6孔板中,滴加細胞懸液,放入37 ℃、5% CO2培養箱培養,待細胞貼壁后,更換無血清培養基處理24 h,用4%多聚甲醛溶液室溫固定15 min,加3‰ TritonX-100 37 ℃孵育10 min,用10%山羊血清配制的兔抗大鼠Kv1.5、Kv4.3、Cav1.2、Cx40和Cx43 抗體4 ℃過夜。次日以山羊抗兔IgG-FITC 37 ℃孵育1 h,DAPI復染細胞核5 min,用抗熒光淬滅劑封片后,于倒置熒光顯微鏡下觀察并攝片。采用Image J軟件分析各組熒光圖片的平均熒光強度(median fluorescent intensity, MFI)值。
2.5 Real-time PCR檢測mRNA的表達 收集各組細胞,采用TRIzol法按提取細胞總RNA,逆轉錄為cDNA。參照GenBank設計并合成引物,GAPDH的上游引物序列為5’-GGGCCAAAAGGGTCATCATCTC-3’,下游引物序列為5’-GCCCTTCCACGATGCCAAA-3’;Kv1.5的上游引物序列為5’-GGGGCAAGATCGTGGGTTC-3’,下游引物序列為5’-CCTGCTCCTCGTGGTCTGTCT-3’;Kv4.3的上游引物序列為5’-CTGCCCTTCTCAACCCCAAATACTC-3’,下游引物序列為5’-ACCGCTTCAAAACACCAGGACTC-3’;Cav1.2的上游引物序列為5’-TCACCCCCAGCAGCTACTCATC-3’,下游引物序列為5’-CTGCGGGTCTCATCTGGAAACATA-3’;Cx40的上游引物序列為5’-GCACACTGTGCGCATGCAGG-3’,下游引物序列為5’-TGCTGGCCTTACTAAGGCG-3’;Cx43的上游引物序列為5’-ATCCAGTGGTACATCTATGG-3’,下游引物序列為5’-CTGCTGGCTCTGCTGGAAGG-3’。反應條件為:95 ℃ 60 s;95 ℃ 10 s、60 ℃ 30 s,共40個循環。以GAPDH為內參照,目的基因mRNA的表達水平用2-ΔΔCt計算。
2.6 Western blot檢測蛋白的表達 收集各組細胞,加入含PMSF的RIPA裂解液提取蛋白,用BCA蛋白濃度測定試劑盒進行定量。取30 μg蛋白,經SDS-PAGE分離后,電轉至PVDF膜上,5%脫脂奶粉室溫封閉90 min,分別加入兔抗大鼠Kv1.5、Kv4.3、Cav1.2、Cx40和Cx43單克隆抗體,4 ℃過夜,加入HRP 標記的山羊抗兔IgG,室溫孵育90 min,TBST 洗膜后ECL 顯影。應用Quantity One 4.6.2凝膠電泳分析軟件進行圖像分析,以目的蛋白與GAPDH蛋白條帶積分光密度的比值進行半定量分析。
3 統計學處理
數據采用SPSS 21.0軟件進行統計分析。計量資料用均數±標準差(mean±SD)表示,多組均數比較采用單因素方差分析(one-way ANOVA),組間兩兩比較應用Bonferroni校正的t檢驗,以P<0.05為差異有統計學意義。
結 果
1 心肌細胞培養與鑒定
細胞培養24 h后,經倒置顯微鏡觀察,大部分細胞已貼壁,成梭形、多邊形,部分細胞出現自發性搏動。培養48 h后呈多邊形,三角形、梭形與不規則形細胞伸出偽足,呈聚集性生長,連接成片,搏動頻率40~150次/分。使用心肌特異性標記的抗橫紋肌α-actinin抗體鑒定心肌細胞,可見心肌細胞純度達95%以上,見圖1。

Figure 1.The images of the cell morphology (A, ×400) and the identification of primary cultured cardiomyocytes with immunofluorescence staining (B, ×200).
2 T3與依普利酮對心肌細胞存活率的影響
CCK-8法測定結果顯示100 nmol/L T3處理組的心肌細胞存活率明顯低于對照組(P<0.05),而1、2、5、10 nmol/L T3組與對照組比較,差異無統計學顯著性。10-5mol/L依普利酮處理組的心肌細胞存活率明顯低于對照組(P<0.05),而10-8、10-7、10-6mol/L T3組與對照組比較,差異無統計學顯著性。據此結果選擇10 nmol/L為T3處理濃度,10-6mol/L為依普利酮的處理濃度,見圖2。

Figure 2.The concentration-response relationship of T3 and eplerenone on the viabilities of the cardiomyocytes by CCK-8 assay. Mean±SD. n=3. *P<0.05 vs 0 nmol/L (T3); #P<0.05 vs 0 mol/L (Epl).
3 免疫熒光法評價T3與依普利酮對Kv1.5、Kv4.3、Cav1.2、Cx40和Cx43的蛋白相對表達水平的影響
與對照組比較,T3組的Kv1.5、Kv4.3、Cav1.2與Cx40的MFI升高(P<0.01),Cx43的MFI降低(P<0.01),Epl組的Kv1.5、Kv4.3、Cav1.2與Cx40的MFI降低(P<0.01),Cx43的MFI升高(P<0.01)。與T3組比較,T3+Epl組的Kv1.5、Kv4.3、Cav1.2與Cx40的MFI降低(P<0.01),Cx43的MFI升高(P<0.01),見圖3。
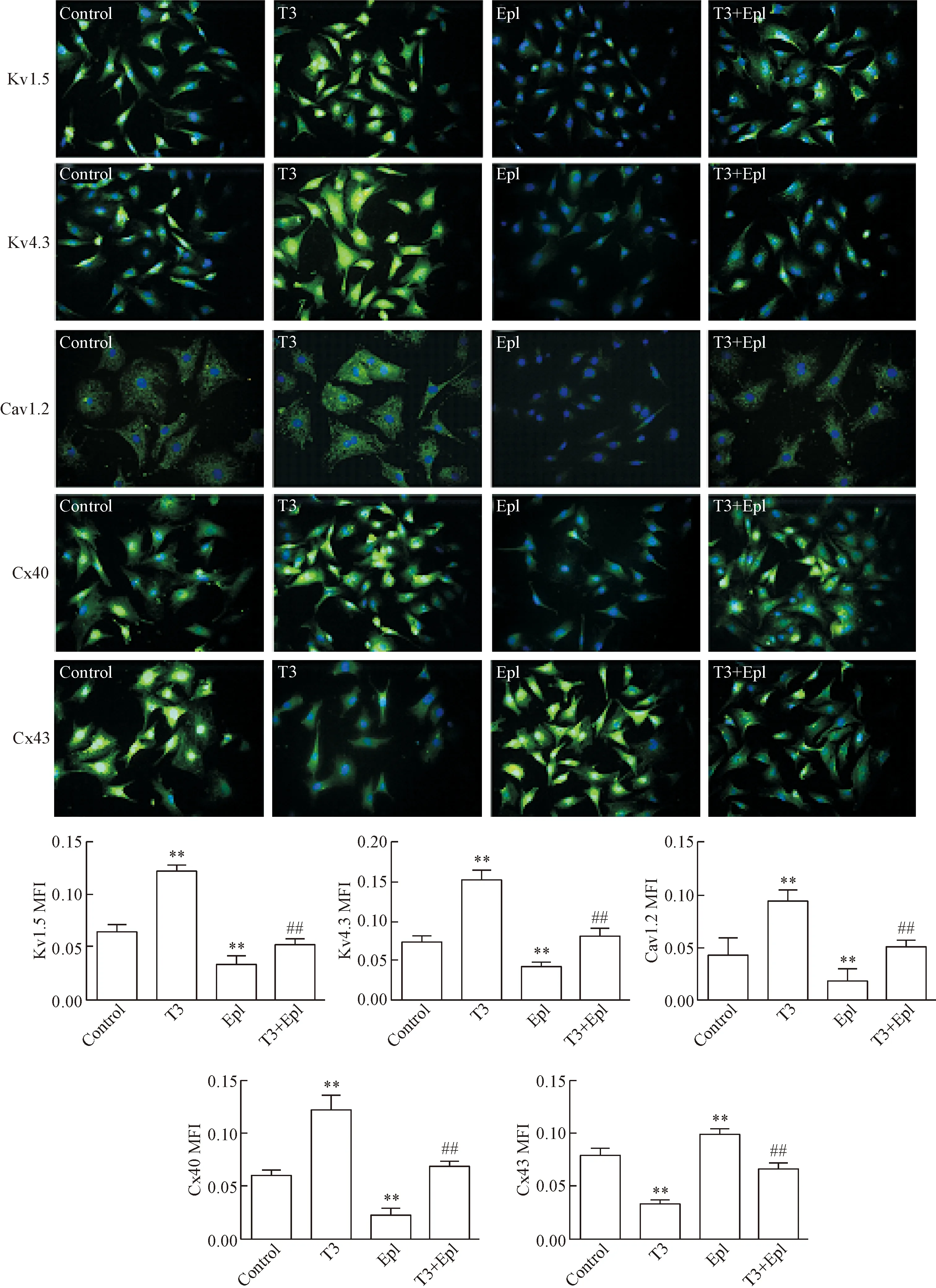
Figure 3.The effects of T3 and eplerenone on the protein expression of Kv1.5, Kv4.3, Cav1.2, Cx40 and Cx43 in the cardiomyocytes by immunofluorescence staining (×400). Mean±SD. n=5. **P<0.01 vs control; ##P<0.01 vs T3.
4 T3與依普利酮對Kv1.5、Kv4.3、Cav1.2、Cx40和Cx43 mRNA表達的影響
T3、依普利酮干預24 h后,與對照組比較,T3組的Kv1.5、Cav1.2與Cx40的mRNA水平分別增加了2.61、2.01和2.44倍(P<0.01),Kv4.3的mRNA水平增加了1.31倍(P<0.05),Cx43的mRNA水平減少了63%(P<0.01)。Epl組Kv1.5和Kv4.3的mRNA水平分別減少了51%和48%(P<0.05),而Cav1.2與Cx40的mRNA水平降低與對照組比較差異無統計學顯著性,Cx43的mRNA水平則增加了1.45倍(P<0.01)。與T3組比較,T3+Epl組Kv1.5、Kv4.3、Cav1.2與Cx40的mRNA表達降低(P<0.01), Cx43的mRNA表達升高(P<0.01),見圖4。
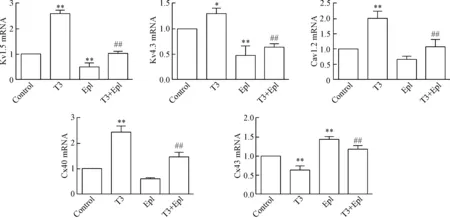
Figure 4.The mRNA expression of Kv1.5, Kv4.3, Cav1.2, Cx40 and Cx43 in the cardiomyocytes detected by real-time PCR. Mean±SD. n=3. *P<0.05, **P<0.01 vs control; ##P<0.01 vs T3.
5 T3與依普利酮對Kv1.5、Kv4.3、Cav1.2、Cx40和Cx43蛋白表達的影響
與對照組比較,T3組的Kv1.5、Kv4.3、Cav1.2與Cx40 蛋白表達均升高,Cx43 蛋白表達降低,Epl組的Kv1.5、Kv4.3、Cav1.2與Cx40 蛋白表達均降低,Cx43的蛋白表達升高。與T3組比較,T3+Epl組的Kv1.5、Kv4.3、Cav1.2與Cx40蛋白表達均降低,Cx43的蛋白表達升高,見圖5。
討 論
本研究證實甲狀腺激素可誘導心肌細胞Kv1.5、Kv4.3、Cav1.2、Cx40和Cx43的表達發生改變,提示甲狀腺素可導致心肌細胞電重構。既往研究已證實醛固酮拮抗劑可以改善心肌纖維化等結構重構,而對于電重構相關研究則較少。本研究結果提示醛固酮拮抗劑依普利酮可通過抑制Cav1.2、Kv1.5、Kv4.3、Cx40的mRNA及蛋白表達上調與Cx43的表達下調,在一定程度上改善甲狀腺激素導致的心肌電重構。
ICa-L是心肌細胞重要的去極化電流,在動作電位2相開放,內流的Ca2+促進肌漿網釋放Ca2+,觸發心肌收縮偶聯。心肌電重構時,進入細胞內Ca2+增多,并誘發肌漿網釋放更多Ca2+,使細胞內Ca2+異常升高,細胞內鈣超負荷,觸發鈣依賴式通道失活,使L型鈣通道活性降低,L型鈣離子通道Cav1.2電流密度下降,動作電位時程和有效不應期縮短,從而促進多環折返的形成,這是Cav1.2的mRNA及蛋白表達下降導致心律失常的可能機制[5-6]。L型鈣通道Cav1.2的表達上調亦可導致細胞內鈣超負荷,導致心肌電重構[7]。本研究發現在T3處理心肌細胞24 h后L型鈣通道Cav1.2的mRNA及蛋白表達上調,依普利酮能逆轉T3導致的Cav1.2上調。
鉀離子流是心肌細胞動作電位復極的主要外向電流。Kv1.5與Kv4.3 分別是心肌細胞超快延遲IKur與Ito形成的基礎。本研究結果提示,心肌細胞在T3干預后Kv1.5與Kv4.3 mRNA的表達分別升高2.61和1.31倍,且蛋白表達呈一致升高,表明在甲狀腺激素誘導的心肌細胞中,鉀電流可能由于Kv1.5與Kv4.3 的變化而發生變化,從而引起心肌細胞動作電位時程的改變,導致心肌電重構。有研究發現過表達Kv4.3可增加Ito電流密度,縮短動作電位時程,減少Ca2+內流[8-9]。因此,鉀離子通道Kv4.3可通過調節跨膜Ca2+內流,引起L型鈣通道變化,這可能是Kv4.3導致心肌電重構的原因之一。
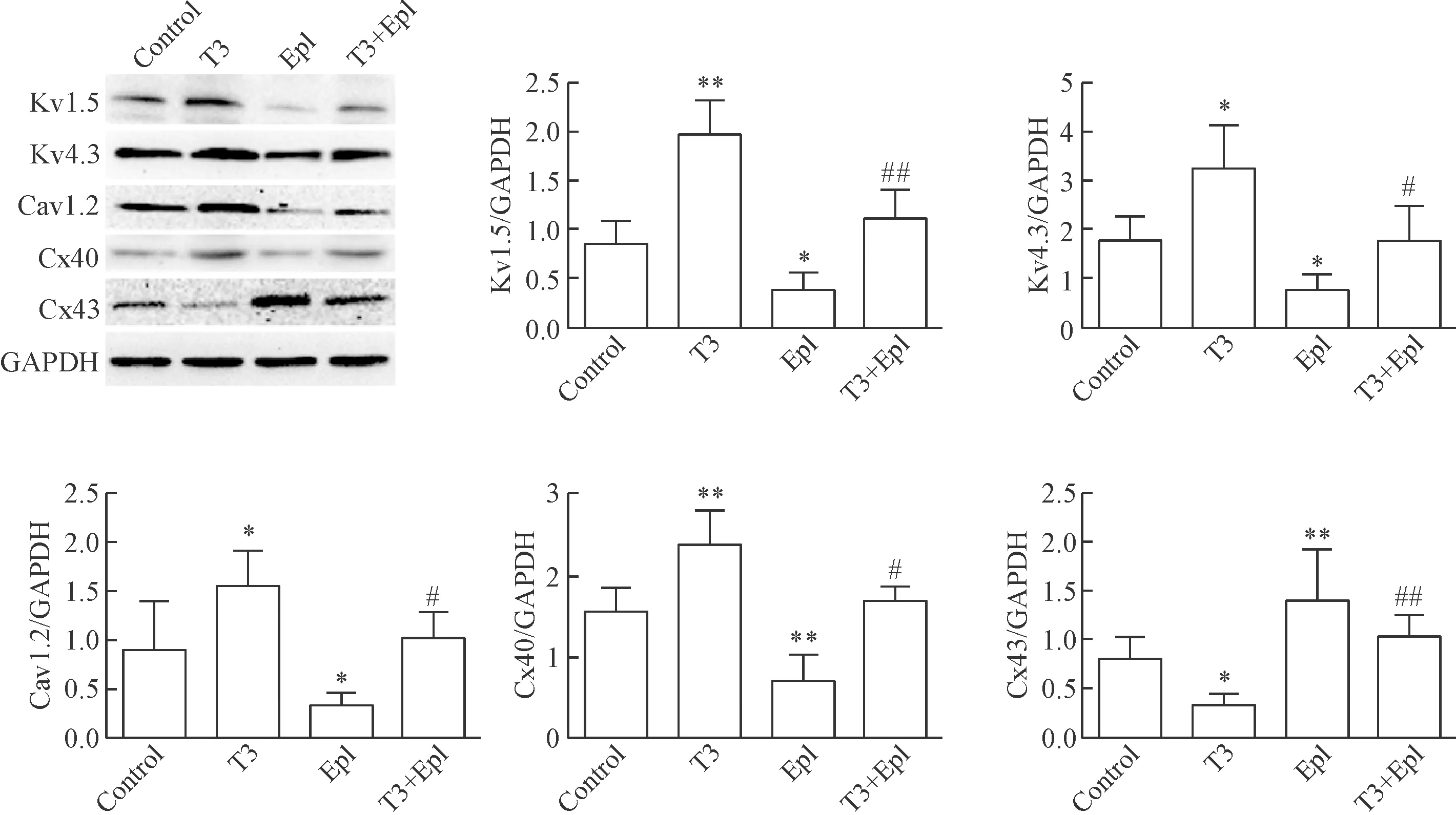
Figure 5.The protein expression of Kv1.5, Kv4.3, Cav1.2, Cx40 and Cx43 in the cardiomyocytes determined by Western blot. Mean±SD. n=5~7. *P<0.05, **P<0.01 vs control; #P<0.05, ##P<0.01 vs T3.
縫隙連接蛋白在心肌細胞表面組成縫隙連接通道,是心肌細胞之間進行電化學信息交換的分子基礎,其結構和功能的異常與心肌電重構的發生關系密切。Cx40主要分布于心房肌及浦肯野傳導系統中,而Cx43廣泛分布于心房肌及心室肌細胞,Cx40與Cx43是心房電激動傳導的關鍵蛋白。本研究發現T3誘導的心肌細胞中Cx43 mRNA和蛋白的表達下調并伴有Cx40 mRNA和蛋白的表達上調,與Dupont 等[10]在心肌肥大患者心內膜表面發現的結果一致。但有關心肌電重構Cx43的表達存在爭議,Cx43可在AngⅡ誘導的心肌細胞中的表達上調[11],在壓力超負荷犬的心肌中未見異常,但其分布不均[12],而在甲亢大鼠中表達降低[13]。本研究結果提示,T3可使Cx43的表達下調,而Epl可部分逆轉這一效應。研究結果不一致的原因不甚清楚,可能與不同的干預條件有關。結合國內外多項研究結果,我們推測,無論Cx43的表達升高或降低或分布異常,均打亂了縫隙連接通道蛋白的平衡,干擾了心肌細胞電化學的正常傳導,導致了心肌細胞的電重構。
甲亢時心肌組織中AngⅡ水平升高。本課題組的前期研究也證實,甲狀腺素誘導后心肌組織的AngⅡ水平升高,表明高甲狀腺素誘導的心肌電重構中,有RAAS系統的參與[14]。醛固酮拮抗劑依普利酮可以通過鹽皮質激素受體抑制醛固酮的活性。研究發現醛固酮拮抗劑依普利酮可通過激活生電鈉泵,改善心肌纖維化,減少心臟結構重構及心律失常的發生[15],同時可通過調節肌漿網Ca2+-ATP酶的表達改善心衰大鼠的心肌電重構[16]。本研究結果顯示,T3誘導的心肌細胞Kv1.5、Kv4.3、Cav1.2、Cx40和Cx43的改變可被依普利酮逆轉,進一步提示醛固酮拮抗劑依普利酮可在一定程度上改善甲狀腺激素誘導的心肌電重構。因此,醛固酮拮抗劑可能通過調節離子通道蛋白和縫隙連接蛋白表達而具有抗心律失常作用,我們將在后續的研究中,以全細胞膜片鉗技術進一步證實其對上述離子通道電生理學功能的影響。
[1] Nattel S, Harada M. Atrial remodeling and atrial fibrillation: recent advances and translational perspectives[J]. J Am Coll Cardiol, 2014, 63(22):2335-2345.
[2] Dai DZ. Vulnerable substrate and multiple ion channel disorder in a diseased heart will be new targets for antiarrhythmic therapy[J]. Acta Pharmacol Sinica, 2000, 21(4):289-295.
[3] Mayyas F, Alzoubi KH, Van Wagoner DR. Impact of aldosterone antagonists on the substrate for atrial fibrillation: aldosterone promotes oxidative stress and atrial structural/electrical remodeling[J]. Int J Cardiol, 2013, 168(6):5135-5142.
[4] Yang J, Young MJ. Mineralocorticoid receptor antagonists-pharmacodynamics and pharmacokinetic differences[J]. Curr Opin Pharmacol, 2016, 27:78-85.
[5] Chen WJ, Yeh YH, Lin KH, et al. Molecular characterization of thyroid hormone-inhibited atrial L-type calcium channel expression: implication for atrial fibrillation in hyperthyroidism[J]. Basic Res Cardiol, 2011, 106(2):163-174.
[6] Dartsch T, Fischer R, Gapelyuk A, et al. Aldosterone induces electrical remodeling independent of hypertension[J]. Int J Cardiol, 2013, 164(2):170-178.
[7] Yu Z, Wang T, Xu L, et al. Thyroid hormone increased L-type calcium channel mRNA expression and L-type calcium current of myocytes in rabbits[J]. Biomed Mater Eng, 2012, 22(1-3):49-55.
[8] Kaprielian R, Wickenden AD, Kassiri Z, et al. Relationship between K+channel down-regulation and [Ca2+]iin rat ventricular myocytes following myocardial infarction[J]. J Physiol, 1999, 517(Pt 1):229-245.
[9] Lebeche D, Kaprielian R, Hajjar R. Modulation of action potential duration on myocyte hypertrophic pathways[J]. J Mol Cell Cardiol, 2006, 40(5):725-735.
[10]Dupont E, Matsushita T, Kaba RA, et al. Altered connexin expression in human congestive heart failure[J]. J Mol Cell Cardiol, 2001, 33(2):359-371.
[11]Teunissen BE, Jongsma HJ, Bierhuizen MF. Regulation of myocardial connexins during hypertrophic remodelling[J]. Eur Heart J, 2004, 25(22):1979-1989.
[12]Vetter C, Zweifel M, Zuppinger C, et al. Connexin 43 expression in human hypertrophied heart due to pressure and volume overload[J]. Physiol Res, 2010, 59(1):35-42.
[13]Marchlewska K, Kula K, Walczak-Jedrzejowska R, et al. Maturational changes in connexin 43 expression in the seminiferous tubules may depend on thyroid hormone action[J]. Arch Med Sci, 2013, 9(1):139-145.
[14]Kobori H, Hayashi M, Saruta T. Thyroid hormone stimulates renin gene expression through the thyroid hormone response element[J]. Hypertension, 2001, 37(1):99-104.
[15]De Mello WC. Beneficial effect of eplerenone on cardiac remodelling and electrical properties of the failing heart[J]. J Renin Angiotensin Aldosterone Syst, 2006, 7(1):40-46.
[16]Kobayashi N, Yoshida K, Nakano S, et al. Cardioprotective mechanisms of eplerenone on cardiac performance and remodeling in failing rat hearts[J]. Hypertension, 2006, 47(4):671-679.
(責任編輯: 林白霜, 余小慧)
Eplerenone partly reverses thyroid hormone induced myocardial electrical remodeling
ZHAO Lu, YI Tian-tian, LUO Shui-lian, SU Li
(DepartmentofCardiology,TheSecondAffiliatedHospitalofChongqingMedicalUniversity,Chongqing400010,China.E-mail:sulicq@163.com)
AIM: To investigate the effects of eplerenone (Epl) on thyroid hormone (T3) induced myocardial electrical remodeling. METHODS:The ventricles of 1~3 d neonatal rats were digested with 0.125% trypsin and 0.08% collagenase type 2. The cell suspension was replated for 90 min to reduce the proportion of non-myocardial cells. The isolated cardiomyocytes were randomly divided into control group, T3 group, Epl group and T3+Epl group. The cardiomyocytes were identified by immunofluorescence staining. The viability of the cardiomyocytes was measured by CCK-8 assay. The expression of Kv1.5, Kv4.3, Cav1.2, connexin 40 (Cx40) and Cx43 at mRNA and protein levels was determined by immunofluorescence staining, real-time PCR and Western blot. RESULTS:The results of the cell immunofluorescence labeling conformed that the cultured cells were cardiomyocytes with more than 95% positive staining of sarcomeric α-actinin. Compared with control group, the mRNA and protein levels of Kv1.5, Kv4.3, Cav1.2 and Cx40 were increased, but the expression of Cx43 was decreased in T3 group. The mRNA and protein levels of Kv1.5, Kv4.3, Cav1.2 and Cx40 were decreased, but the expression of Cx43 was increased in Eplerenone group. Compared with T3 group, the mRNA and protein expression levels of Kv1.5, Kv4.3, Cav1.2 and Cx40 were decreased, but the expression of Cx43 was increased in T3+Epl group. CONCLUSION: Eplerenone partly reverses T3-induced myocardial electrical remodeling.
Cardiomyocytes; Thyroid hormone; Eplerenone; Ion channel; Connexin
1000- 4718(2017)03- 0428- 07
2016- 09- 26
2016- 11- 15
△通訊作者 Tel: 023-63693452; E-mail: sulicq@163.com
R363
A
10.3969/j.issn.1000- 4718.2017.03.008
