蒺藜苜蓿、天藍苜蓿、金花菜基因組SNP穿梭標記開發
2017-04-14 08:03:04任海龍魏臻武陳祥揚州大學動物科學與技術學院江蘇揚州225009新疆農業科學院海南三亞農作物育種試驗中心海南三亞57204
草業學報 2017年4期
任海龍,魏臻武,陳祥(.揚州大學動物科學與技術學院,江蘇 揚州 225009;2.新疆農業科學院海南三亞農作物育種試驗中心,海南 三亞 57204)
?
蒺藜苜蓿、天藍苜蓿、金花菜基因組SNP穿梭標記開發
任海龍1,2,魏臻武1*,陳祥1
(1.揚州大學動物科學與技術學院,江蘇 揚州 225009;2.新疆農業科學院海南三亞農作物育種試驗中心,海南 三亞 572014)
蒺藜苜蓿是繼擬南芥和水稻之后又一個進行全基因組測序的植物,利用蒺藜苜蓿的基因組序列,開發出可以在其他豆科植物上應用的分子標記,即穿梭標記,已成為缺乏基因組信息或基因組復雜的豆科植物基因組學及分子遺傳學研究的有效手段。天藍苜蓿和金花菜是我國最重要的兩種一年生苜蓿,由于缺乏有效的分子標記,這兩種苜蓿在基因組水平上的研究很少。SLAF-seq是近年來開發出的一種簡化基因組測序技術,具有高通量、準確性、成本低、周期短的優點,已在眾多物種的全基因組SNP標記開發上得到應用。本研究通過SLAF-seq技術對12份蒺藜苜蓿、天藍苜蓿和金花菜材料進行簡化基因組測序,共得到28.04×106個讀長的測序數據,276432個高質量的SLAF標簽,其中58748個SLAF標簽為多態性標簽,平均測序深度為17.44。在58748個多態性SLAF標簽中,共檢測出次要基因型頻率(MAF)大于0.05的SNP標記189133個。本研究開發出的SNP標記可用于一年生苜蓿的遺傳多樣性、遺傳圖譜構建和重要農藝性狀的QTL定位等的研究,其種間穿梭的特性可為苜蓿屬種間基因組排列順序、系統進化關系、比較圖譜構建等方面的研究提供幫助。
蒺藜苜蓿;天藍苜蓿;金花菜;SLAF;SNP
苜蓿屬(Medicago)大約有87個種,包括了最重要的豆科牧草——紫花苜蓿(Medicagosativa)和豆科模式植物——蒺藜苜蓿(Medicagotruncatula),在農業和畜牧業生產中占有重要地位[1]。按照生育期的長短,苜蓿屬植物可分為一年生苜蓿(annual medic)和多年生苜蓿(perennial medic),其中大部分為一年生苜蓿[2]。一年生苜蓿被廣泛應用于歐洲和澳大利亞的草地農業系統,但是國內對一年生苜蓿的研究卻很少。
蒺藜苜蓿是繼擬南芥和水稻之后又一個進行全基因組測序的植物[3],因其具有基因組小(500 Mb)、遺傳轉化效率高、生長周期短、自花授粉等優點,使之成為豆科生物學和基因組學研究的模式植物[4]。相對于另一種豆科模式植物——百脈根(Lotuscorniculatus),蒺藜苜蓿與許多重要豆科作物的親緣關系更近,表現出更好的共線性關系,被認為是豆科比較基因組學研究的重要工具[5]。利用蒺藜苜蓿的基因組序列,開發出可以在其他豆科植物上應用的分子標記,即穿梭標記,已成為缺乏基因組信息或基因組復雜的豆科植物基因組學及分子遺傳學研究的有效手段[6-8]。天藍苜蓿和金花菜是我國分布最廣泛的兩種一年生苜蓿[4]。天藍苜蓿(Medicagolupulina)在全世界廣泛分布,除用于建植草地收獲飼草外,更多用于冬季裸露土地的覆蓋等生態保護上。因其匍匐生長和侵占性強的特點,天藍苜蓿還可以與其他作物間作套種,起到肥土和抑制雜草的作用[9-10]。現遍及我國東北、華北、西北和云貴高原,生態類型十分豐富[11]。金花菜(Medicagopolymorpha)起源于地中海和相鄰的干旱、半干旱地區,由于其低光周期敏感性和春化性而廣泛分布于世界各地[12]。除能固定大氣中的N2,生產出高質量的牧草外[13],還具有土壤修復、培肥地力、防止水土流失等作用,被廣泛應用于牧草、飼料、綠肥、草地農業系統、冬季覆蓋作物和救荒牧草等領域[14-16]。除此之外,金花菜還有很多優良的農藝性狀:如土壤和氣候的適應性[17]、持久性、良好的冬季生長能力和耐放牧等[18],可用于過度放牧或焚燒后退化草場的恢復[16],被認為是苜蓿改良的重要遺傳資源[19]。現在我國的江浙地區和云南的栽培面積較大。目前,除蒺藜苜蓿外,其他一年生苜蓿的研究主要還是集中在耕作制度、栽培措施和生產應用上,基因組水平上的研究很少,缺少有效的分子標記是其中主要的原因之一。
單核苷酸多態性(single nucleotide polymorphism,SNP)是近年來發展起來的第三代分子標記。具有數量多、分布廣、高通量、易于分型和自動化等特點,被廣泛應用于分子遺傳學的各個領域[20-21]。得益于下一代測序(next-generation sequencing,NGS)技術的飛速發展,全基因組尺度的海量SNP標記開發已成為可能[22]。但是,基于重測序技術的SNP標記開發費用較高,并且僅限于已經完成全基因組測序的物種[21]。因此,以部分序列代替全基因組序列的簡化基因組測序技術得到迅速發展。其中SLAF-seq(specific length amplification fragment sequencing)[23]技術因其具有通量高、準確性高、成本低、周期短的特點,已在眾多作物的SNP標記開發上得到應用[24-27]。本研究利用 SLAF-seq技術,以1份蒺藜苜蓿、1份天藍苜蓿和10份金花菜資源為材料,通過限制性酶切片段的雙端測序,獲得理想的SLAF標簽,進而開發高質量的SNP穿梭標記,為基于SNP標記的一年生苜蓿系統進化關系、遺傳多樣性、遺傳圖譜構建、重要農藝性狀的QTL定位和比較基因組學等研究工作奠定基礎。
1 材料與方法
1.1 試驗材料
試驗材料為來自不同地區的3種一年生苜蓿,分別為蒺藜苜蓿材料1份,天藍苜蓿材料1份和金花菜材料10份,其中金花菜材料來自于我國5個省份或直轄市,詳細信息見表1。以上材料均由揚州大學草業科學研究所提供。
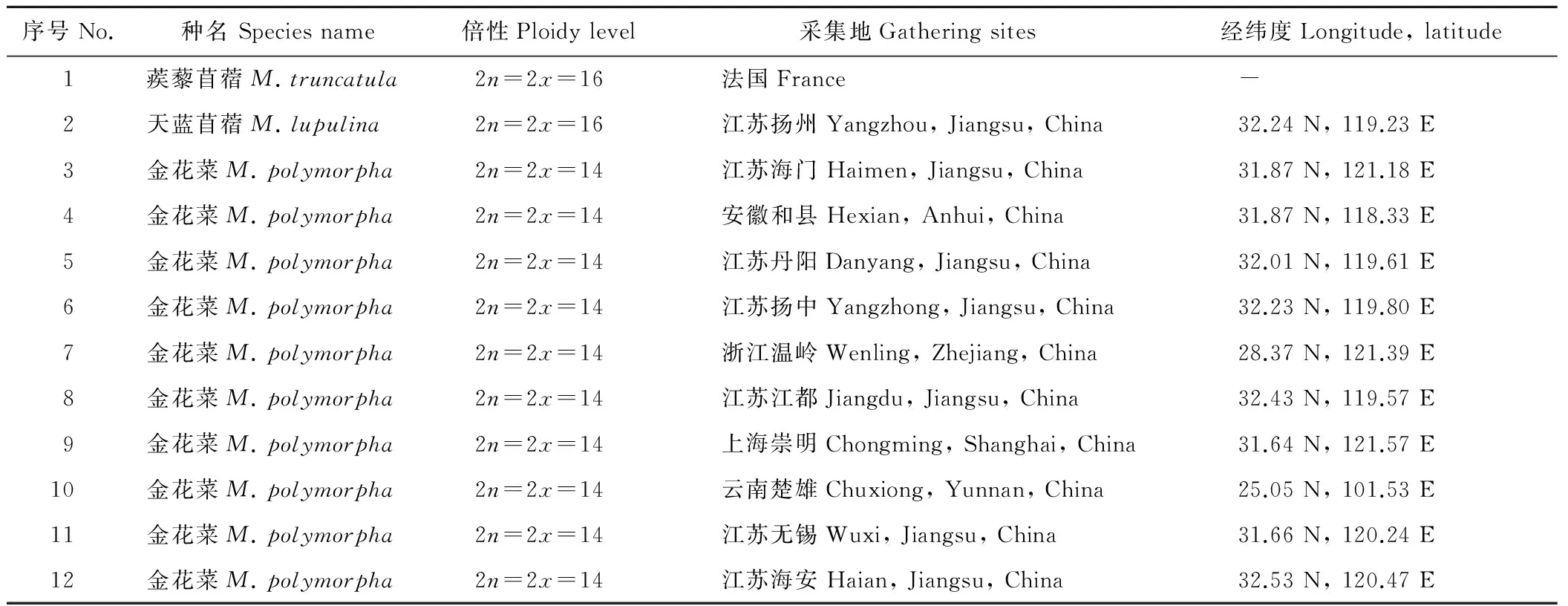
表1 供試一年生苜蓿名稱與來源Table 1 Name and origin of tested annual medics
1.2 田間種植情況
試驗地位于江蘇省揚州市揚州大學揚子津東校區(東經119°26′,北緯32°23′),土壤為沙壤土,肥力中等。12份一年生苜蓿材料于2014年10月進行播種,12月采集葉片,翌年4月完成收獲。播種前每份材料都經過了至少2代單粒傳繁育,以保證試驗材料的純度。
1.3 基因組DNA 的提取與檢測
一年生苜蓿的DNA提取參照改良的CTAB法[28]。提取的DNA濃度和質量用紫外分光光度計ND-1000(NanoDrop, Wilmington, DE, USA)和1%的瓊脂糖凝膠電泳檢測。
1.4 SLAF文庫構建及測序
以蒺藜苜蓿的基因組為參考基因組,利用北京百邁客生物科技有限公司研發的酶切預測軟件進行酶切預測,確定最適的酶切方案。酶切方案需滿足以下四點:1)位于重復序列的酶切片段比例盡可能低;2)酶切片段在基因組上盡量均勻分布;3)酶切片段長度與實驗體系的吻合程度高;4)最終獲得酶切片段(SLAF標簽)數滿足實驗要求。
根據選定的最適酶切方案,對檢測合格的各樣品基因組DNA分別進行酶切實驗。對得到的酶切片段(SLAF標簽)進行3′端加A處理、連接Dual-index測序接頭、PCR擴增、純化、混樣、切膠選取目的片段,文庫質檢合格后用IlluminaHiSeq TM 2500 進行 PE125 bp測序,測序數據已提交到NCBI數據庫 (索取號:SRP082533)。為評估建庫實驗的準確性,選用擬南芥(Arabidopsisthaliana)的基因組作為對照(Control),進行相同的處理參與建庫和測序。
1.5 SLAF標簽的獲得和SNP標記的開發
利用Dual-index對測序得到的原始數據進行識別,得到各個樣品的讀長數據。對過濾完接頭的測序讀長進行測序質量和數據量的評估。通過擬南芥(control)數據的比對效率來評估酶的酶切效率,判斷實驗過程的準確性和有效性。根據序列相似性將各樣品的讀長進行聚類,聚類到一起的讀長來源于一個SLAF片段(SLAF標簽)。由于同一SLAF標簽在不同材料間的序列相似度高于同一材料不同SLAF標簽的相似度,所以根據序列相似度可確定不同的SLAF標簽。同一個SLAF標簽在不同樣品間序列有差異即可定義為多態性的SLAF標簽。再根據多態性SLAF標簽序列上的差異進行SNP標記的開發。
2 結果與分析
2.1 SLAF建庫評估
通過對蒺藜苜蓿基因組進行電子酶切預測, 確定了RsaⅠ和HaeⅢ的雙酶切方案為最適的SLAF標簽建庫酶切方案,該方案的酶切片段長度為314~414 bp,預測到了117528個SLAF標簽,這些SLAF標簽在基因組上分布均勻,位于重復序列區域的SLAF標簽比例僅為6.19%(表2)。本實驗的SLAF建庫以相同條件處理下的擬南芥為對照,擬南芥酶切片段的雙端比對效率為83.41%,酶切效率為97.79%(表3),說明RsaⅠ和HaeⅢ的雙酶切實驗對3種一年生苜蓿酶切片段的建庫效率很高。
2.2 測序數據及SLAF標簽分析
經過文庫構建和測序,共獲得28.04×106個讀長數據(表4)。其中,蒺藜苜蓿獲得了1560664個讀長數據;天藍苜蓿獲得了1643954個讀長數據,金花菜材料的讀長數據范圍在1584219個到5157030個之間,平均每個材料產生2483115個讀長數據。測序質量值(Q)是評估高通量測序單堿基錯誤率的重要指標,測序質量值越高對應的堿基測序錯誤率越低。如果某堿基測序出錯的概率為0.001,則該堿基的質量值Q應該為30。本實驗測序質量值Q30的范圍在83.43%至86.40%之間,均值為85.14%,且所有樣品的Q30值均在80%以上,說明測序堿基錯誤率低,所獲測序數據合格。測序數據的GC含量范圍在37.35%至39.03%,均值為 38.22%,GC含量普遍不高,說明達到測序要求。
通過對測序片段的聚類分析,本次實驗共開發得到276432個SLAF標簽(表5)。其中,蒺藜苜蓿獲得了100007個SLAF標簽, 天藍苜蓿獲得了100085個SLAF標簽,金花菜獲得的SLAF標簽數量在104196到116918之間,高于前兩種一年生苜蓿,這與金花菜參試材料的數量較多有關。另外,測序深度的增加也會一定程度提高同質材料中SLAF標簽的開發數量。本實驗中一年生苜蓿材料SLAF標簽的測序深度在12.04到36.97之間,平均測序深度為17.44,其中金花菜材料“溫嶺”和“楚雄”達到了30以上,說明本次實驗的測序質量較高。

表2 酶切預測確定的酶切方案信息統計Table 2 Information prediction according to the enzyme digestion method
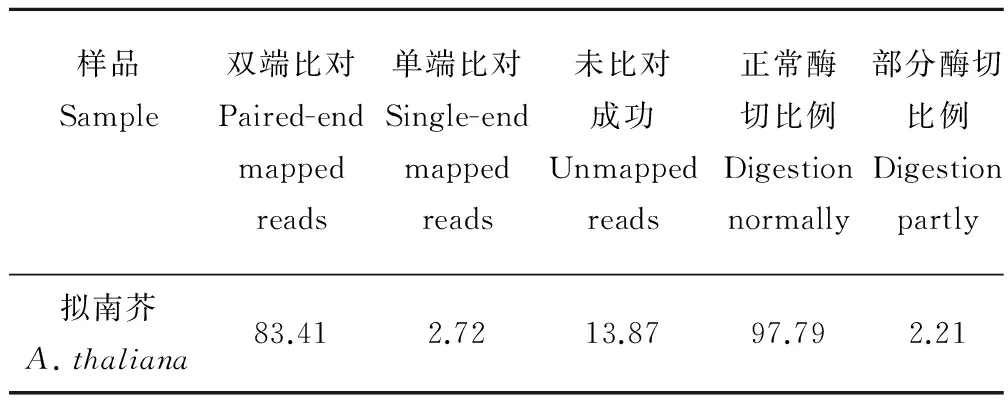
表3 擬南芥測序讀長比對結果Table 3 The alignment results between obtained reads and its genome sequences in Arabidopsis %
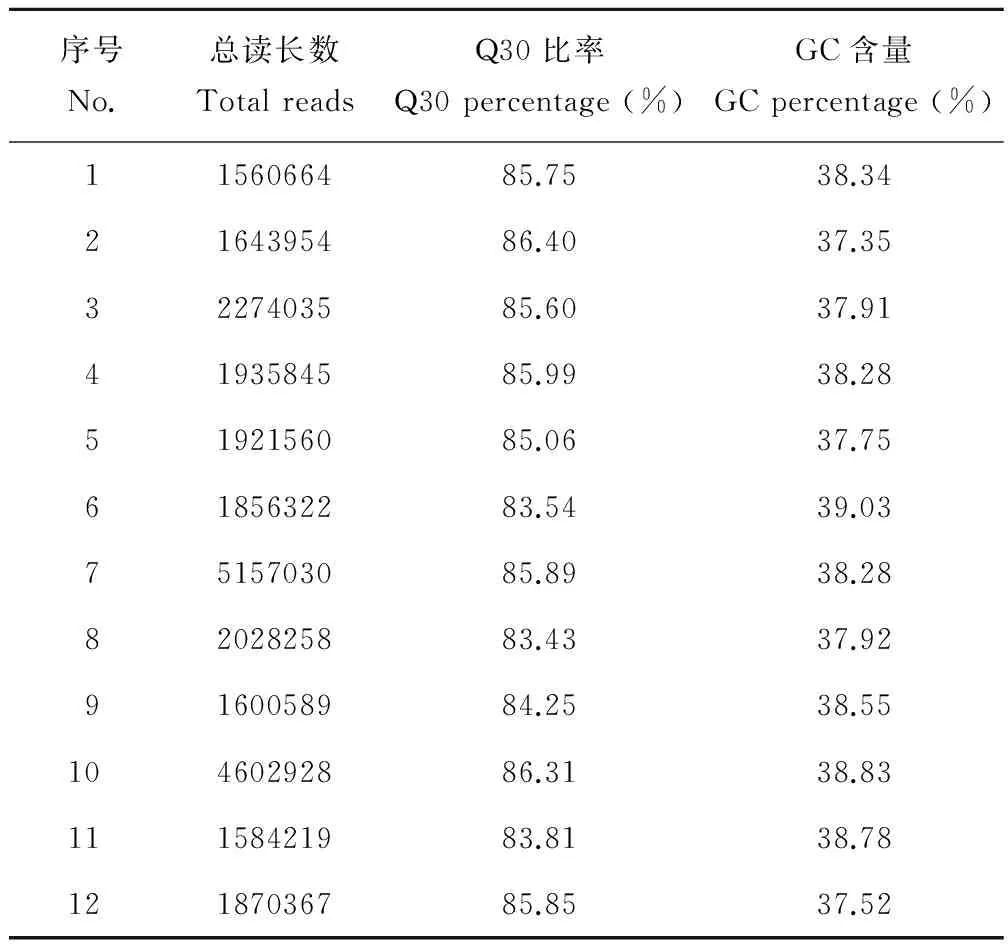
表4 測序質量評估結果Table 4 Quality assessment of sequencing
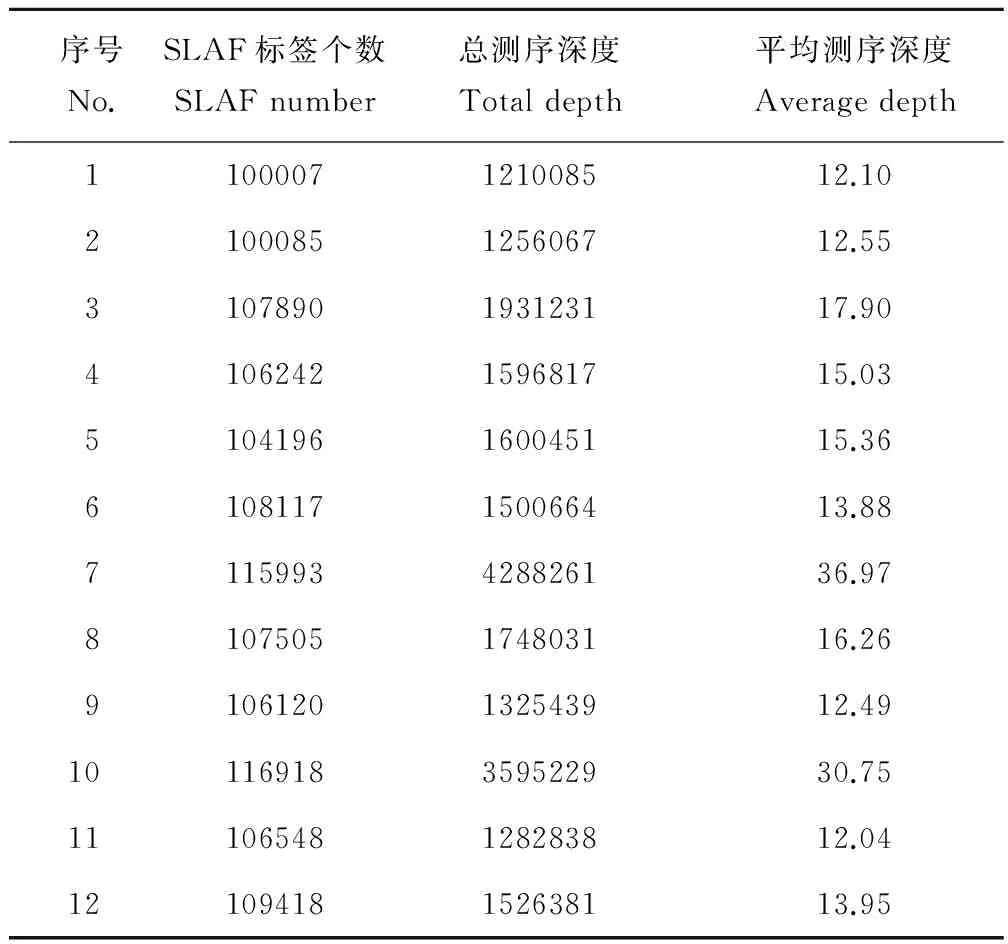
表5 各材料SLAF標簽信息統計Table 5 Evaluation of SLAF for each accession
獲得的全部276432個SLAF標簽可以分成多態性標簽、非多態性標簽和重復性標簽3種類型(表6)。其中多態性SLAF標簽共計58748個,占全部SLAF標簽數量的21.25%,可用于后續SNP標記的開發。

表6 不同類型SLAF數量統計Table 6 Different types of SLAF tags
2.3 基于SLAF標簽的SNP 標記開發
根據開發得到的58748個多態性SLAF標簽的信息,按照次要基因型頻率(MAF)>0.05 進行篩選,共得到 189133個高質量的種間SNP穿梭標記。其中蒺藜苜蓿獲得了89433個SNP標記,天藍苜蓿獲得81203個SNP標記,金花菜材料獲得了130054個到154110個不等的種間SNP標記(表7)。
從表7中可以看出,蒺藜苜蓿和天藍苜蓿的SNP完整度,即SNP的數量要明顯低于金花菜。由于12份一年生苜蓿材料的SLAF標簽數量差別不大,因此造成SNP標記數量差異的原因可能與苜蓿種間多態性SLAF標簽的數量和突變類型不同有關。表明材料間親緣關系的遠近會影響SLAF-seq技術開發SNP標記的數量,材料間親緣關系越遠,開發出的SNP標記數量越少。此外,SNP的數量和測序深度也有一定關系,來自“溫嶺”和“楚雄” 金花菜材料的平均測序深度是其他金花菜材料的2.32倍,SNP標記的開發數量也相應增加了17.18%。
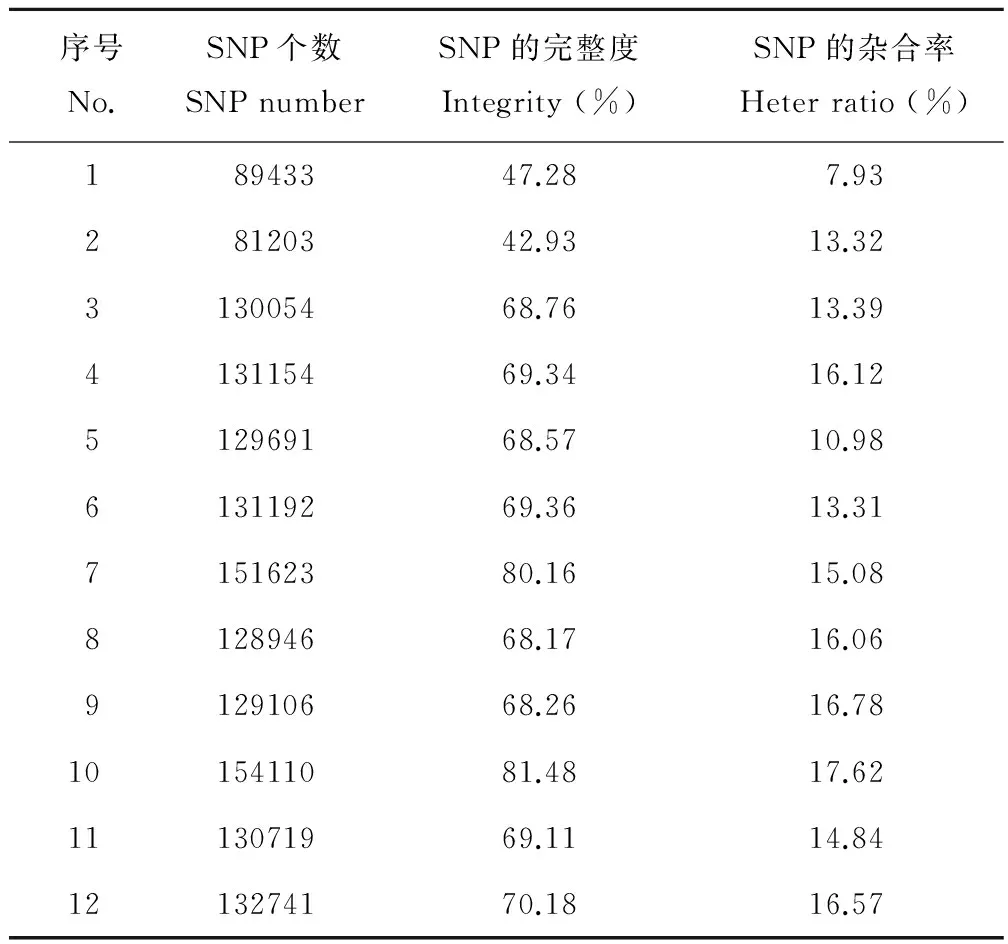
表7 各材料SNP標簽信息統計Table 7 Evaluation of SNP for each accession
3 討論
3.1 SLAF-seq技術SNP標記開發的可行性
SLAF-seq技術是以生物信息學為基礎的簡化基因組測序技術。包括通過模擬酶切確定酶切方案,酶切片段的雙端測序,對照基因組的測序質量評估,SLAF標簽的獲得等步驟,可在全基因組尺度上進行海量SNP標記的開發。因其具有通量高、準確性高、成本低、周期短等優勢,已在多種作物上取得良好效果。如Zhang等[24]通過SLAF-seq策略構建了第一張芝麻高密度遺傳圖譜,圖譜中93.03%的標記類型是SNP標記;Li等[25]運用SLAF-seq技術,將5785個SNP標記成功整合到了一張由SLAF標簽構建的大豆遺傳圖譜上,并利用這些標記對異黃酮含量相關基因進行了QTL定位;Wei等[26]運用同樣策略,構建了一張由1800個SNP標記組成的黃瓜遺傳圖譜,并對黃瓜果實長度、重量等性狀進行了基因定位。最近,蘇文瑾等[27]通過SLAF-seq技術從300份甘薯種質資源中獲得了795794個高質量SNP標記,為后續甘薯的遺傳學研究奠定了基礎。眾多研究表明,SLAF-seq技術已成為大規模SNP標記開發的有效方法。本研究通過SLAF-seq技術,成功地開發了189133個覆蓋蒺藜苜蓿、天藍苜蓿和金花菜3種一年生苜蓿全基因組的SNP穿梭標記,為一年生苜蓿大規模的分子標記開發提供了可行策略。
3.2 SNP標記在一年生苜蓿上的應用展望
3.2.1 在系統進化關系研究上的應用前景 苜蓿屬植物種間和種內遺傳變異巨大,且存在種間雜交現象,造成了苜蓿屬植物分類的困擾和爭議[29]。傳統的分類學為認識和利用苜蓿屬植物提供了幫助,但也帶來了一定程度的混亂。例如,部分胡盧巴屬植物與苜蓿屬植物具有相同的彈花機制,使兩屬間的界限十分模糊[30];依賴莢果螺旋形態分類,又將黃花苜蓿(Medicagofalcata)和雜交苜蓿排除在苜蓿屬之外[31];糙邊苜蓿(Medicagomurex)同時具有2n=14和2n=16兩種染色體類型,紫花苜蓿同時具有二倍體和四倍體,按照染色體數目的分類也行不通。Yoder等[32]的研究認為全基因組水平的分子標記可為苜蓿屬植物,特別是一年生苜蓿的分類和種屬間親緣關系的確定提供可行策略。毫無疑問,作為基因組中最廣泛的變異形式,SNP標記將為一年生苜蓿的系統進化研究提供幫助。
3.2.2 在遺傳多樣性研究上的應用前景 一年生苜蓿在表型和農藝性狀上存在著豐富的遺傳變異,在過去的幾十年中,科研人員一直十分重視其遺傳多樣性的研究。以金花菜為例,1994年Bullitta等[12]利用12個生化標記,對意大利撒丁島不同氣候和土壤條件下的45份金花菜自然群體進行了遺傳變異情況的分析;隨后,Hannachi等[33]采用6個酶標記對來自突尼斯的16份金花菜資源也進行了類似的研究;2000年Paredes等[34-35]利用12個同工酶標記研究了智利41個金花菜自然群體的遺傳多樣性,并進一步用40對RAPD(random amplification polymorphic DNA)標記對其中36份金花菜資源進行了驗證。最近,Chu等[36]利用從蒺藜苜蓿基因組中開發的5對SSR(simple sequence repeats)標記,對來自中國8個金花菜個體進行了遺傳多樣性研究。然而,傳統分子標記的數量有限,在作物遺傳多樣性的研究中難免力不從心,甚至得到不可靠的結論。數以萬計的SNP標記,將為一年生苜蓿遺傳多樣性的研究提供可靠手段。
3.2.3 在遺傳圖譜構建上的應用前景 遺傳圖譜是進行QTL定位、基因圖位克隆以及分子標記輔助育種的基礎。蒺藜苜蓿的遺傳圖譜已經成為豆科作物重要農藝性狀基因定位的重要工具[37]。然而,由于缺乏有效的分子標記,其他一年生苜蓿尚未有遺傳圖譜的報道。2014年,Li等[38]利用另一種簡化基因組測序技術(genotyping-by-sequencing,GBS),成功將3591個SNP標記鉚釘在2130 cM的紫花苜蓿遺傳圖上,構建了迄今為止密度最高的紫花苜蓿遺傳圖譜,證明了SNP標記在苜蓿遺傳圖譜應用上的巨大潛力。一年生苜蓿多為純合二倍體,相對于雜合四倍體紫花苜蓿,利用SNP標記構建遺傳圖譜將更為簡單有效。
3.2.4 SNP穿梭標記的應用前景 穿梭標記是比較基因組學研究的重要工具,利用穿梭標記可以對近緣物種間的基因組結構、基因組排列順序等進行比較分析,發掘物種進化過程中的基因組片斷缺失、倍增、倒位以及轉位等信息,還可以與模式物種建立比較圖譜,實現在基因組信息缺乏的物種中克隆重要的功能基因[39]。作為模式植物,從蒺藜苜蓿基因組中開發出的分子標記已在眾多豆科作物中得到應用[6-8,36]。蒺藜苜蓿與天藍苜蓿和金花菜同屬一年生苜蓿,親緣更近[40],由此推斷蒺藜苜蓿與天藍苜蓿和金花菜的比較基因組學研究將更為有效。
4 結論
本研究通過SLAF-seq技術對12份蒺藜苜蓿、天藍苜蓿和金花菜材料進行簡化基因組測序,共得到28.04×106個讀長的測序數據,276432個高質量的SLAF標簽,其中58748個SLAF標簽為多態性標簽,平均測序深度為17.44。在58748個多態性SLAF標簽中,共檢測出189133個高質量的SNP穿梭標記。本研究開發出的SNP穿梭標記,將為今后一年生苜蓿的系統發生學、遺傳多樣性、遺傳圖譜構建和重要農藝性狀的QTL定位及比較基因組學等研究提供幫助。
References:
[1] Steele K P, Ickert-Bond S M, Zarre S,etal. Phylogeny and character evolution inMedicago(Leguminosae): Evidence from analyses of plastid trnK/matK and nuclear GA3ox1 sequences. American Journal of Botany, 2010, 97(7): 1142-1155.
[2] Brummer E C, Bouton J H, Kochert G. Analysis of annualMedicagospecies using RAPD markers. Genome, 1995, 38(2): 362-367.
[3] Chen A M, Lian R L, Sun J,etal. Leguminous model plant-Medicagotruncatula. Plant Physiology Communications, 2006, 42(5): 997-1003. 陳愛民, 連瑞麗, 孫杰, 等. 豆科模式植物——蒺藜苜蓿. 植物生理學報, 2006, 42(5): 997-1003.
[4] Wei Z W, Gai J Y. Model legume:Medicagotruncatula. Acta Prataculturae Sinica, 2008, 17(1): 114-120. 魏臻武, 蓋鈞鎰. 豆科模式植物——蒺藜苜蓿. 草業學報, 2008, 17(1): 114-120.
[5] Young N D, Cannon S B, Sato S,etal. Sequencing the genespaces ofMedicagotruncatulaandLotusjaponicus. Plant Physiology, 2005, 137(4): 1174-1181.
[6] Choi H K, Kim D, Uhm T,etal. A sequence-based genetic map ofMedicagotruncatulaand comparison of marker colinearity withM.sativa. Genetics, 2004, 166(3): 1463-1502.
[7] Gupta S, Prasad M. Development and characterization of genic SSR markers inMedicagotruncatulaand their transferability in leguminous and non-leguminous species. Genome, 2009, 52(9): 761-771.
[8] Gupta D, Taylor P W J, Inder P,etal. Integration of EST-SSR markers ofMedicagotruncatulainto intraspecific linkage map of lentil and identification of QTL conferring resistance to ascochyta blight at seedling and pod stages. Molecular Breeding, 2012, 30(1): 429-439.
[9] Feng Y Q, Cao Z Z. Advanced studies onMedicagolupulinautilization. Pratacultural Science, 2005, 22(2): 16-20. 馮毓琴, 曹致中. 天藍苜蓿栽培利用的研究進展. 草業科學, 2005, 22(2): 16-20.
[10] Hively W D, Cox W J. Interseeding cover crops into soybean and subsequent corn yields. Agronomy Journal, 2001, 93(2): 308-313.
[11] Feng Y Q, Cao Z Z, Jin J F. Analysis on Correlation Traits Affecting Single Yield ofMedicagolupulinaL[C]. Hohhot: The Second Conference of The Chinese Society of Grass and The International Symposium, 2004. 馮毓琴, 曹致中, 金巨芳. 影響天藍苜蓿單株產量諸因素的相關性分析[C]. 呼和浩特: 中國草學會二次會議暨國際學術研討會, 2004.
[12] Bullitta S, Floris R, Hayward M D,etal. Morphological and biochemical variation in Sardinian populations ofMedicagopolymorphaL. suitable for rainfedMediterraneanconditions. Euphytica, 1994, 77(3): 263-268.
[13] Khaef N, Sadeghi H, Taghvaei M. Effects of new strategies for breaking dormancy of two annual medics (MedicagoscutellataandMedicagopolymorpha). American-Eurasian Journal of Agricultural and Environmental Sciences, 2011, 11: 626-632.
[14] Denton M D, Hill C R, Bellotti W D,etal. Nodulation ofMedicagotruncatulaandMedicagopolymorphain two pastures of contrasting soil pH and rhizobial populations. Applied Soil Ecology, 2007, 35(2): 441-448.
[15] Nichols P G H, Loi A, Nutt B J,etal. New annual and short-lived perennial pasture legumes for Australian agriculture-15 years of revolution. Field Crops Research, 2007, 104(1): 10-23.
[16] Graziano D, Di Giorgio G, Ruisi P,etal. Variation in pheno-morphological and agronomic traits among burr medic (MedicagopolymorphaL.) populations collected in Sicily, Italy. Crop and Pasture Science, 2010, 61(1): 59-69.
[17] Ewing M A, Robson A D. The effect of nitrogen supply on the early growth and nodulation of several annualMedicagospecies. Crop and Pasture Science, 1990, 41(3): 489-497.
[18] Loi A, Howieson J G, Cocks P S,etal. The adaptation ofMedicagopolymorphato a range of edaphic and environmental conditions: effect of temperature on growth, and acidity stress on nodulation and nod gene induction. Animal Production Science, 1993, 33(1): 25-30.
[19] Scarpa G M, Pupilli F, Damiani F,etal. Plant regeneration from callus and protoplasts inMedicagopolymorpha. Plant Cell, Tissue and Organ Culture, 1993, 35(1): 49-57.
[20] Wang J, Luo M C, Chen Z,etal. Aegilops tauschii single nucleotide polymorphisms shed light on the origins of wheat D-genome genetic diversity and pinpoint the geographic origin of hexaploid wheat. New Phytologist, 2013, 198(3): 925-937.
[21] Kumar S, Banks T W, Cloutier S. SNP discovery through next-generation sequencing and its applications. International Journal of Plant Genomics, 2012, doi: 10.1155/2012/831460.
[22] Etter P D, Bassham S, Hohenlohe P A,etal. SNP discovery and genotyping for evolutionary genetics using RAD sequencing. Molecular Methods for Evolutionary Genetics, 2011, 772: 157-178.
[23] Sun X, Liu D, Zhang X,etal. SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS One, 2013, 8(3): e58700.
[24] Zhang Y, Wang L, Xin H,etal. Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplified fragment (SLAF) sequencing. BMC Plant Biology, 2013, 13(1): 141. doi:10.1186/1471-2229-13-141.
[25] Li B, Tian L, Zhang J,etal. Construction of a high-density genetic map based on large-scale markers developed by specific length amplified fragment sequencing (SLAF-seq) and its application to QTL analysis for isoflavone content inGlycinemax. BMC Genomics, 2014, 15(1):1086. doi: 10.1186/1471-2164-15-1086.
[26] Wei Q, Wang Y, Qin X,etal. An SNP-based saturated genetic map and QTL analysis of fruit-related traits in cucumber using specific-length amplified fragment (SLAF) sequencing. BMC Genomics, 2014, 15(1): 1158. doi:10.1186/1471-2164-15-1158.
[27] Su W J, Zhao N, Lei J,etal. SNP sites developed by specific length amplification fragment sequencing (SLAF-seq) in sweet potato. Scientia Agricultura Sinica, 2016, 49(1): 27-34. 蘇文瑾, 趙寧, 雷劍, 等. 基于SLAF-seq技術的甘薯SNP位點開發. 中國農業科學, 2016, 49(1): 27-34.
[28] Murray M G, Thompson W F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Research, 1980, 8(19): 4321-4326.
[29] Lu X S. The exploration and classification of genetic resources of genusMedicagoin China. Chinese Journal of Grassland, 2009, 31(5): 17-22. 盧欣石. 中國苜蓿屬植物遺傳資源分類整理探究. 中國草地學報, 2009, 31(5): 17-22.
[30] Deng C H, Cui D F, Yang H J,etal. Studies on morphological characters and numerical classification ofMedicagoL. andTrigonellaL. species. Journal of Plant Resources and Environment, 2010, 19(4): 1-11. 鄧超宏, 崔大方, 羊海軍, 等. 苜蓿屬和胡盧巴屬植物的形態特征及數量分類研究. 植物資源與環境學報, 2010, 19(4): 1-11.
[31] Lu X S. Status of germplasm classifications for genusMedicago. Acta Agrestia Sinica, 2009, 17(5): 680-685. 盧欣石. 苜蓿屬植物分類研究進展分析. 草地學報, 2009, 17(5): 680-685.
[32] Yoder J B, Briskine R, Mudge J,etal. Phylogenetic signal variation in the genomes ofMedicago(Fabaceae). Systematic Biology, 2013, 62(3): 424-438.
[33] Hannachi A S, Boussaid M, Marrakchi M. Genetic variability organisation and gene flow in natural populations ofMedicagopolymorphaL. prospected in Tunisia. Genetics Selection Evolution, 1998, 30(1): 121-135.
[34] Paredes M, Becerra V, Correa P,etal. Isozymatic diversity in accesions ofMedicagopolymorphacollected along an environmental gradient in Chile, and its relationship with other species ofMedicago. Revista Chilena de Historia Natural, 2000, 73(3): 479-488.
[35] Paredes M, Becerra V, Rojo C,etal. Ecotypic differentiation inMedicagopolymorphaL. along an environmental gradient in central Chile. RAPDs studies show little genetic divergence. Euphytica, 2002, 123(3): 431-439.
[36] Chu H J, Yan J, Hu Y,etal. Cross-species amplification of 92 microsatellites ofMedicagotruncatula. Molecular Ecology Resources, 2010, 10(1): 150-155.
[37] Thoquet P, Ghérardi M, Journet E P,etal. The molecular genetic linkage map of the model legumeMedicagotruncatula: an essential tool for comparative legume genomics and the isolation of agronomically important genes. BMC Plant Biology, 2002, 2(2): 385-391.
[38] Li X, Wei Y, Acharya A,etal. A saturated genetic linkage map of autotetraploid alfalfa (MedicagosativaL.) developed using genotyping-by-sequencing is highly syntenous with theMedicagotruncatulagenome. G3: Genes|Genomes|Genetics, 2014, 4(10): 1971-1979.
[39] Zhao J R. Study on Cross-species Markers Related to Disease Resistance Between Soybean andMedicagotruncatula[D]. Nanning: Guangxi University, 2007. 趙金榮. 大豆與豆科模式物種蒺藜苜蓿間與抗病相關的穿梭標記研究[D]. 南寧: 廣西大學, 2007.
[40] Bena G, Prosperi J M, Lejeune B,etal. Evolution of annual species of the genusMedicago: a molecular phylogenetic approach. Molecular Phylogenetics & Evolution, 1998, 9(3): 552-559.
Cross-species markers developed from genome sequencing inMedicagotruncatula,MedicagolupulinaandMedicagopolymorpha
REN Hai-Long1,2, WEI Zhen-Wu1*, CHEN Xiang1
1.CollegeofAnimalScience&Technology,YangzhouUniversity,Yangzhou225009,China; 2.HainanCenterofXinjiangAcademyofAgriculturalSciences,Sanya572014,China
Medicagotruncatulais another whole genome sequenced species next toArabidopsisthalianaandOryzasativa. Studying genomics and genetics using cross-species markers developed fromM.truncatulahas become an important strategy for species with more complex genomes or for legumes with less well understood genome.M.lupulinaandM.polymorphaare the most important annual medics in China. However, limited by molecular marker, little research has been done on genome scales in these species. Specific length amplification fragment sequencing (SLAF-seq) possesses significant advantages including development of markers with high throughput, high accuracy, low cost and time saving which has been successfully used in single nucleotide polymorphism (SNP) development in many species. In this study, 28.04×106reads were obtained from a collection of twelve ofM.truncatula,M.lupulinaandM.polymorphaaccessions using SLAF-seq. The average sequencing depth was 17.44, and 276432 high-quality SLAFs were developed, among which 58748 SLAFs were polymorphic. Further 189133 cross-species SNPs were identified from these polymorphic SLAFs with minor allele frequencies (MAFs)>0.05. These SNPs can be used in genetic diversity, genetic linkage mapping and QTL mapping of important agronomic traits in annual medic. They will also convey benefits for the study of genome orders, system evolution, and comparative map construction.
Medicagotruncatula;Medicagolupulina;Medicagopolymorpha; SLAF; SNP
10.11686/cyxb2016400
http://cyxb.lzu.edu.cn
2016-11-01;改回日期:2016-12-26
江蘇省科技支撐計劃項目(BE2012340)和新疆維吾爾自治區公益性科研院所基本科研業務費項目(KYGY2016121)資助。
任海龍(1985-),男,黑龍江阿城人,助理研究員,在讀博士。E-mail: renhailong_2006@163.com*通信作者Corresponding author. E-mail: zhenwu_wei@hotmail.com
任海龍, 魏臻武, 陳祥. 蒺藜苜蓿、天藍苜蓿、金花菜基因組SNP穿梭標記開發. 草業學報, 2017, 26(4): 188-195.
REN Hai-Long, WEI Zhen-Wu, CHEN Xiang. Cross-species markers developed from genome sequencing inMedicagotruncatula,MedicagolupulinaandMedicagopolymorpha. Acta Prataculturae Sinica, 2017, 26(4): 188-195.
