分散固相萃取結合液相色譜-串聯(lián)質譜法測定飼料中8種脂溶性著色劑
2017-04-27 06:04:32吳銀良
分析測試學報 2017年4期
付 巖,朱 勇,吳銀良
(寧波市農業(yè)科學研究院,浙江 寧波 315040)
分散固相萃取結合液相色譜-串聯(lián)質譜法測定飼料中8種脂溶性著色劑
付 巖,朱 勇,吳銀良*
(寧波市農業(yè)科學研究院,浙江 寧波 315040)
建立了飼料中8種脂溶性著色劑(對位紅、蘇丹紅Ⅰ、蘇丹紅Ⅱ、蘇丹紅Ⅲ、蘇丹紅Ⅳ、蘇丹紅7B、蘇丹紅G、蘇丹黃)含量的液相色譜-串聯(lián)質譜測定方法。飼料樣品中脂溶性著色劑經乙腈提取,離心后上清液采用分散固相萃取凈化,凈化液稀釋后進行LC-MS/MS分析。樣品測定時采用Acquity BEH C18色譜柱進行色譜分離,以0.2%甲酸溶液-乙腈作為流動相進行梯度洗脫,電噴霧正離子(ESI+)模式電離,多反應監(jiān)測(MRM)模式檢測,同位素稀釋內標法定量。8種脂溶性著色劑在1.0~200 μg/L范圍內線性關系良好,相關系數(r2)均大于0.998;在飼料中的方法檢出限為5.0 μg/kg,定量下限為10 μg/kg。在10,50,500 μg/kg加標濃度下8種脂溶性著色劑的回收率為102%~111%,批內相對標準偏差(RSD)為2.8%~8.0%,批間RSD為2.8%~7.8%。該方法能滿足飼料樣品中脂溶性著色劑監(jiān)控的需要。
分散固相萃取;飼料;脂溶性著色劑;液相色譜-串聯(lián)質譜法;殘留
蘇丹紅染料(Sudan dyes)是非生物合成親脂性偶氮化合物,對位紅(Para red)是一種常用的紅色顏料,亦稱對硝苯胺紅,屬于偶氮系列化工合成染色劑。蘇丹紅染料和對位紅主要應用于機油、蠟、油彩、汽油等工業(yè)產品[1-2]。研究表明對位紅對眼睛、皮膚和呼吸系統(tǒng)有刺激性[3];蘇丹紅Ⅰ~Ⅳ被國際癌癥研究中心列為三級致癌物,蘇丹紅Ⅲ初級代謝產物 4-氨基偶氮苯、蘇丹紅Ⅳ初級代謝產物鄰氨基偶氮甲苯和鄰甲基苯胺均列為二級致癌物,對人可能致癌[4]。因而很多國家目前禁止蘇丹紅染料和對位紅作為食品添加劑使用。雖然合理合規(guī)使用色素為促進我國飼料工業(yè)發(fā)展作出了一定貢獻,但由于有些畜禽養(yǎng)殖企業(yè)或個人不遵守國家法律法規(guī)濫用色素,或非法使用未經批準的有毒工業(yè)合成顏料,給我國的動物性食品安全造成了一定的問題。因此為保障飼料和食品安全,我國農業(yè)部于2007年制定了農業(yè)行業(yè)標準NY/T 1258-2007《飼料中蘇丹紅染料的測定 高效液相色譜法》[5]。然而該標準中脂溶性色素僅包括蘇丹紅Ⅰ~Ⅳ,未包括與之性質相近的對位紅和其它蘇丹紅染料;同時利用該標準測定時,在靈敏度和準確性方面不如普遍使用的液相色譜-串聯(lián)質譜法(LC-MS/MS),因此從保障我國人民身體健康出發(fā),建立簡單、快速的測定飼料中包括蘇丹紅在內的脂溶性色素的分析方法具有重要意義。
目前蘇丹紅染料的檢測方法常使用高效液相色譜法[6-7]和液相色譜-串聯(lián)質譜法[8-10],主要集中在飼料[11]及雞蛋[12]、辣椒[13]等食品,這些方法通常僅對蘇丹紅Ⅰ~Ⅳ進行分析,同時分析與之性質相近的對位紅和其它蘇丹紅染料的報道較少[14-15]。在前處理手段上,常結合凝膠滲透色譜法[16]和固相萃取技術對樣品進行進一步凈化,如采用C18固相萃取柱、免疫親和柱、中性氧化鋁小柱等固相萃取柱進行凈化[17-19],但這些方法工序復雜、操作繁瑣,成本較高,而使用分散固相萃取,具有簡便、快速、廉價且凈化效果良好的特點。此外,使用同位素內標法定量可使結果更加準確,但目前已發(fā)表的方法中采用同位素內標稀釋法進行定量分析的較少[20-21]。本研究針對現有方法存在的前處理復雜、成本高等問題,采用分散固相萃取,通過優(yōu)化樣品前處理及色譜-質譜條件,建立了分散固相萃取/高效液相色譜-串聯(lián)質譜測定配合飼料(豬、雞、魚)中8種脂溶性著色劑的方法。該法可同時測定的藥物種類多,靈敏度高,提取凈化效果好,簡化了前處理步驟,降低了成本。同時采用同位素內標法定量,有效消除了基質效應,結果準確可靠,為風險監(jiān)測提供了技術支持。
1 實驗部分
1.1 儀器與試劑
Waters UPLC XevoTMTQ MS超高效液相色譜-串聯(lián)質譜儀(美國Waters公司),配置電噴霧離子源;IKA Vortex Genie 3旋渦振蕩器(德國),低溫高速離心機(德國Sigma公司)。
脂溶性著色劑標準品:蘇丹紅Ⅰ、蘇丹紅Ⅱ、蘇丹紅Ⅲ、蘇丹紅Ⅳ、對位紅、蘇丹紅7B、蘇丹紅G、蘇丹黃的純度均大于95%,購自德國 Dr.Ehrenstorfer GmbH;脂溶性著色劑同位素標記標準品:蘇丹紅Ⅰ-D5、蘇丹紅Ⅱ-D6、蘇丹紅Ⅲ-D6、蘇丹紅Ⅳ-D6、蘇丹紅G-D3、蘇丹黃-D5的純度均大于95%,購自德國 Dr.Ehrenstorfer GmbH;乙腈(色譜純,德國Merck公司);甲酸(色譜純,美國Tedia公司);PSA(40~60 μm,美國Agilent公司),實驗用水為 Milli-Q 超純水。
1.2 樣品前處理
1.2.1 提 取 稱取飼料樣品5 g(精確至0.01 g),置于50 mL聚四氟乙烯塑料離心管中,準確加入10 μg/mL的脂溶性著色劑同位素標記標準中間液100 μL,混合15 s,加入25 mL乙腈,于恒溫振蕩器上振蕩提取30 min,離心,取上清液備用。
1.2.2 凈 化 準確吸取上清液2 mL移入含50 mg PSA填料的帶蓋10 mL塑料離心管中,渦旋30 s后,以5 000 r/min離心3 min,取0.85 mL上清液與0.15 mL 0.2%甲酸溶液混合,過0.22 μm有機濾膜后供上機測定。
1.3 色譜-質譜條件
色譜柱:Acquity BEH C18(100 mm×2.1 mm,1.7 μm);流動相:A相為0.2%甲酸溶液,B相為乙腈;梯度洗脫條件:B相在0.5 min內保持85 %后,在2.5 min內線性增至95 %,保持1.5 min,然后在0.1 min內降至85 %,保持0.9 min;流速0.30 mL/min;進樣量10 μL。
ESI源正離子模式電離;多反應監(jiān)測(MRM);毛細管電壓:3.8 kV;萃取錐孔電壓:20 V;RF透鏡電壓:0.5 V;離子源溫度:150 ℃;脫溶劑氣溫度:500 ℃;錐孔氣流速:50 L/h;脫溶劑氣流速:1 000 L/h;其它條件詳見表1。

表1 8種脂溶性染料及其內標物的母離子、子離子、錐孔電壓及碰撞能量Table 1 Precursor ions,product ions,cone voltages and collision energies of 8 fat soluble colorants and their internal standards
* quantitative ion
1.4 加標回收率實驗
加標回收率實驗樣品處理同“1.2”,加標濃度為10,50,500 μg/kg,內標物的濃度為200 μg/kg。稱樣后加入標準溶液,渦旋混勻放置0.5 h后進行樣品處理。
2 結果與討論
2.1 質譜條件的優(yōu)化
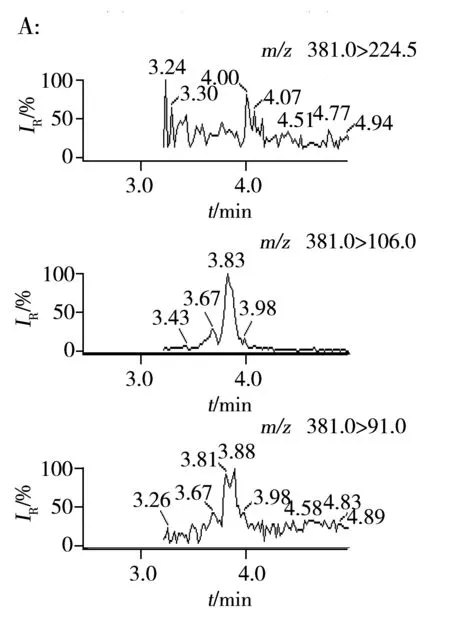

圖1 空白飼料中蘇丹紅Ⅳ 3個子離子(A)及蘇丹紅Ⅱ-D62個子離子(B)的MRM色譜圖Fig.1 MRM chromatograms of sudan Ⅳ(A) and sudan Ⅱ-D6(B) in blank feed
由于蘇丹紅和對位紅分子中含有氮原子,易帶正電荷,因此選擇ESI+離子模式。為優(yōu)化質譜條件,配制含8種著色劑濃度均為100 ng/mL的標準溶液(同時配制相同濃度的6種內標標準溶液)。利用該標準溶液在全掃描方式下,優(yōu)化毛細管電壓、錐孔電壓、裂解溫度、脫溶劑氣流速等參數,得到待測物最強的分子離子峰。 在上述質譜條件下,對選定的母離子進行子離子掃描,優(yōu)化碰撞能量等參數。再經儀器自帶Intellistart軟件優(yōu)化和空白樣品測定后,確定8種脂溶性著色劑和內標物的質譜條件(見表1)。其中蘇丹紅Ⅳ根據文獻報道[13,16]及自帶Intellistart軟件優(yōu)化后起初選擇m/z381.0>91.0和m/z381.0>106.0兩個離子對,但在分析時,m/z381.0>106.0離子對有一定的干擾峰存在(圖1A),對低濃度(添加10 μg/kg)樣品的定性定量會造成較大誤差,因此本文使用靈敏度能達到要求且無干擾的m/z381.0>224.5離子。對于6個內標物質標準溶液采用同樣的方法優(yōu)化質譜條件,優(yōu)化后蘇丹紅Ⅱ-D6起初選擇的是m/z283.0>120.9,但經試驗結果發(fā)現,所有飼料樣品中該離子均存在一定的干擾(圖1B),因此本實驗最后選擇靈敏度能達到要求且無干擾的m/z283.0>162.0離子。
2.2 色譜條件的優(yōu)化
采用乙腈-水為流動相,由于蘇丹紅分子結構中含有偶氮基團,為了防止色譜峰拖尾和提高電離效率,在水相中加入甲酸,并參考杜振霞等[22]研究結果選擇水相中添加甲酸的濃度為0.2%。本文嘗試采用等度洗脫進行色譜分析,水相(含0.2%甲酸溶液)-乙腈的比例為5∶95。結果顯示,采用乙腈提取液直接上樣或經N-丙基乙二胺(PSA)凈化后直接上樣時,8種著色劑的抑制基質效應均非常強,如對位紅直接上樣和50 mg PSA凈化后的抑制率分別達80%和50%。為減少基質抑制效應,同時簡化提取凈化步驟,實驗嘗試利用兩方面措施改善基質效應:①用梯度洗脫增加洗脫時間,減少單位時間雜質量;②在上樣液中加入一定的甲酸溶液,減少雜質總量。結果發(fā)現基質效應得到改善,其中對位紅直接上樣和50 mg PSA凈化后的抑制率分別下降至50%和20%,實驗最終確定的梯度洗脫條件見“1.3”。
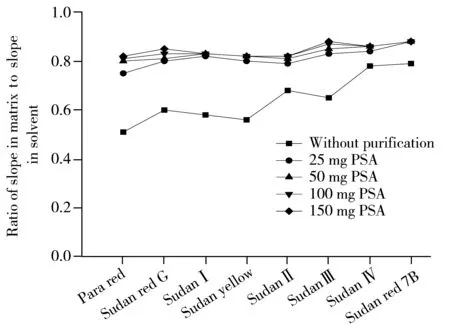
圖2 不同PSA用量下8種脂溶性著色劑的基質效應Fig.2 Matrix effects of eight fat soluble colorants under different amounts of PSA
2.3 前處理條件的選擇
對于食品中的蘇丹紅和對位紅,通常采用在樣品中加入乙腈、正己烷、甲醇、乙醇、二氯甲烷、丙醇、正己烷-丙酮等有機溶劑進行提取,其中前2種溶劑最為常用。而當使用乙腈提取時,溶劑與組織樣品的相容性較好,滲透力強,兼有去蛋白和一定的脫脂功能,同時考慮到方法最終用同位素內標定量,為節(jié)省溶劑用量,采用5 g飼料樣品(加標濃度1.0 mg/kg)考察了25 mL乙腈條件下8種脂溶性色素的提取效率(以外標法定量)。結果顯示,極性越低的色素回收率相對較低,但所有著色劑的回收率均在70%以上,因此最終采用25 mL乙腈對5 g樣品進行提取。經提取后未經基質分散固相萃取凈化及加入不同PSA量凈化的基質效應結果見圖2,從圖2可知未凈化時各著色劑的基質抑制效應較強,當PSA用量達到50 mg及以上時,飼料的基質抑制效應已降到可接受的范圍(斜率比值介于0.8~1.2之間),因此最終確定PSA的用量為50 mg。
2.4 線性實驗
取10 μg/mL混合標準中間液,用0.2%甲酸-乙腈(15∶85)混合溶液逐級稀釋配制濃度分別為1.0,2.0,5.0,20,50,200 μg/L的系列標準工作溶液,每個標準溶液加入100 μL用0.2%甲酸溶液-乙腈(15∶85)稀釋的內標混合工作液(濃度為200 μg/L),使其內標濃度為20 μg/L。LC-MS/MS分析后以定量離子對峰面積和內標峰面積的比值為縱坐標,標準溶液濃度為橫坐標,繪制標準曲線,其中蘇丹紅Ⅰ~Ⅳ、蘇丹黃和蘇丹紅G分別選擇蘇丹紅Ⅰ-D5、蘇丹紅Ⅱ-D6、蘇丹紅Ⅲ-D6、蘇丹紅Ⅳ-D6、蘇丹黃-D5、蘇丹紅G-D3進行定量,對位紅選擇蘇丹紅G-D3進行定量,蘇丹紅7B選擇蘇丹紅Ⅳ-D6進行定量,結果顯示,各著色劑在1.0~200 μg/L范圍內線性關系良好,相關系數(r2)均為0.999。
2.5 方法的回收率、精密度與檢出限
以信噪比S/N=3確定8種脂溶性染料的檢出限為5.0 μg/kg,以信噪比S/N=10確定其定量下限為10 μg/kg。加標回收實驗每批次內同一濃度做5次平行實驗,進行3次重復,因3種配合飼料(豬、雞和魚)的結果基本一致,僅列出豬配合飼料的結果(見表2),相關譜圖見圖3。結果顯示,10~500 μg/kg加標濃度范圍內,方法的平均回收率為102%~111%,批內相對標準偏差(RSD)為2.8%~8.0%,批間RSD為2.8%~7.8%,方法明準確度及精密度能滿足飼料中8種脂溶性著色劑含量的測定要求。

表2 8種脂溶性著色劑的加標回收率及相對標準偏差Table 2 Recoveries and relative standard deviations(RSDs) of 8 fat soluble colorants in feeds
(續(xù)表2)

CompoundAdded(μg/kg)Averagerecovery(%)(n=5)Intra-assayRSD(%)Inter-assayRSD(%)SudanⅣ10,50,500110,105,1037.2,4.9,3.97.7,5.0,4.0Sudanred7B10,50,500111,109,1078.0,6.6,6.57.8,6.3,6.1SudanredG10,50,500105,104,1036.0,4.1,4.75.8,3.9,4.8Sudanyellow10,50,500105,106,1074.0,5.0,5.54.7,5.0,5.3Parared10,50,500104,105,1024.9,4.4,3.94.7,4.9,3.6
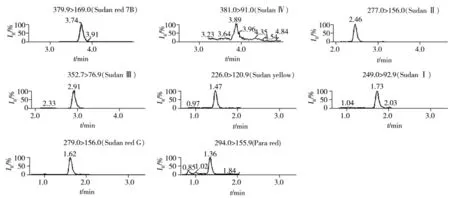
圖3 飼料空白樣品加標10 μg/kg的MRM色譜圖Fig.3 MRM chromatograms of spiked feeds blank sample(10 μg/kg)
2.6 方法應用
采用優(yōu)化后的樣品前處理方法和分析條件對河北省石家莊市獸藥監(jiān)察所提供的陽性飼料樣品進行了分析,其中蘇丹紅Ⅰ有檢出,平均含量為17.3 mg/kg,其他7種脂溶性著色劑均未檢出。
3 結 論
本文建立了飼料中8種脂溶性著色劑同時分析的同位素內標稀釋/液相色譜-串聯(lián)質譜方法。方法簡單快速,樣品經乙腈提取后進行分散固相萃取凈化,凈化液經稀釋后即可進行儀器分析。飼料中8種著色劑在10~500 μg/kg加標濃度范圍內,回收率為102%~111%,批內RSD為2.8%~8.0%,批間RSD為2.8%~7.8%。8種藥物的檢出限均為5.0 μg/kg,定量下限均為10 μg/kg。方法具有準確和靈敏的特點,能滿足飼料中8種藥物殘留量的定量分析要求。
[1] Hahibi M H,Hassanzadch A,Mahdavi S.J.Photochem.Photobiol.A,2005,172(1):89-96.
[2] Nohynek G J,Fautz R,Benech K F,Toutain H.FoodChem.Toxicol.,2004,42(4):517-543.
[3] Zhou X,Li A J,Xu L M,Zhao Q S,Rong H,Chen M Y.FeedRes.(周曉,李愛軍,徐立明,趙慶松,榮會,陳明巖.飼料研究),2007,3:20-23.
[4] Chen Q,Ban G F,Jia Z M,Wu N P,Song Z C,Fang Z Y,Liu S M,Qiu F N.J.Veter.Drug(陳薔,班付國,賈振民,吳寧鵬,宋志超,方忠意,劉素梅,邱富娜.中國獸藥雜志),2009,43(2):24-27.
[5] NY/T 1258-2007.Determination of Sudan Dyes in Feed by HPLC.Agriculture Professional Standard of the People's Republic of China(飼料中蘇丹紅染料的測定 高效液相色譜法.中華人民共和國農業(yè)行業(yè)標準).
[6] Chen N,Liu K F,Zhang Y P.J.Instrum.Anal.(陳娜,劉坤峰,張裕平.分析測試學報),2014,33(8):959-962.
[7] Zhai Y J,Cheng J H.J.Chromatogr.Sci.,2015,53(8):1333-1340.
[8] Wang L,Liu C Y,Wang Z J,Ren X D,Wu D D,You H D,Xiong S.J.Instrum.Anal.(王璐,劉成雁,王志嘉,任雪冬,吳冬冬,尤海丹,熊爽.分析測試學報),2016,35(5):526-531.
[9] Liu Z C,Yang F,Yin T K,Qian J.J.Instrum.Anal.(劉正才,楊方,尹太坤,錢疆.分析測試學報),2015,34(2):171-176.
[10] Li J,Ding X M,Zheng J X,Liu D D,Guo F,Liu H M,Zhang Y B.J.Sep.Sci.,2014,37(17):2439-2445.
[11] Cao Q L,Li R,Fang Z Y,Liu S M.FeedInd.(曹起靈,李銳,方忠意,劉素梅.飼料工業(yè)),2008,29(12):61-62.
[12] Wu N P,Peng L,Ban G F,Jia Z M,Guo F R.Chin.J.Veter.Drug(吳寧鵬,彭麗,班付國,賈振民,郭芙蓉.中國獸藥雜志),2012,46(2):27-29,32.
[13] Huang X H,Liu S Y,Jin Q,Wang S T.Chin.J.HealthLab.Technol.(黃希匯,劉少穎,金銓,王姝婷.中國衛(wèi)生檢驗雜志),2013,23(8):1875-1878.
[14] Qi Y H,Zhang H C,Xu R T,Liu J,Zhang L,Wang J P.J.Environ.Sci.HealthB,2015,50(9):645-653.
[15] Tsaia C F,Kuoa C H,Shihb D Y.J.FoodDrugAnal.,2015,23(3):453-462.
[16] Ban G F,Meng L,Wu N P,Dong Y C,Zhang F W,Du H G,Zhang C W.J.Instrum.Anal.(班付國,孟蕾,吳寧鵬,董穎超,張發(fā)旺,杜紅鴿,張崇威.分析測試學報),2014,33(11):1262-1267.
[17] Wu Y L,Yang T,Zhao J,Huangfu W G,Shen J Z.FoodSci.(吳銀良,楊挺,趙健,皇甫偉國,沈建忠.食品科學),2009,30(16):243-246.
[18] Zhu L X,Meng W,Qiu X M,Liu R R,Xu L,Xu F Y.Adv.Mater.Res.,2013,781/784:1694-1699.
[19] Zhang W G,Yan F,Li D N,Jiang Y,Cao Y,Huang S X.FeedRes.(張文剛,嚴鳳,李丹妮,蔣音,曹瑩,黃士新.飼料研究),2007,5:21-23.
[20] Di W S.Chin.FoodAddit.(邸萬山.中國食品添加劑),2016,3:171-174.
[21] Ning X J,Wang D L,Yu C H,Lu Z Y,Zhang Y Q.J.Chin.MassSpectrom.Soc.(寧嘯駿,王丁林,虞成華,陸志蕓,張燕琴.質譜學報),2009,30(1):41-46.
[22] Du Z X,Sun S Q.Chin.J.Chromatogr.(杜振霞,孫姝琦.色譜),2007,25(5):705-710.
Determination of Eight Fat Soluble Colorants in Feed by Liquid Chromatography-Tandem Mass Spectrometry Combined with Dispersive Solid Phase Extraction
FU Yan,ZHU Yong,WU Yin-liang*
(The Ningbo Academy of Agricultural Sciences,Ningbo 315040,China)
A method was developed for the simultaneous determination of eight fat soluble colorants(para red,sudan Ⅰ,Ⅱ,Ⅲ,Ⅳ,sudan red 7B,sudan red G and sudan yellow) in feed by liquid chromatography-tandem mass spectrometry(LC-MS/MS) combined with dispersive solid phase extraction.The samples were extracted with acetonitrile.Then the extract was purified by dispersive solid phase extraction method with N-propylethylenediamine(PSA) sorbent.After purification,the extract was diluted with 0.2% formic acid solution.Before analysis by LC-MS/MS,the samples were separated on an Acquity BEH C18column with a mixture of 0.2% formic acid solution and acetonitrile as mobile phase under gradient elution conditions.The mass spectrometer was operated under multiple reaction monitoring(MRM) mode in the positive mode.The samples were quantified by the isotope dilution and the internal standard method.Good linearities were obtained for the eight fat soluble colorants in the concentration range of 1.0-200 μg/L with correlation coefficients more than 0.998.The recoveries of the analytes at fortified levels of 10-500 μg/kg were in the range of 102%-111%.The limits of detection and the limit of quantitation for eight fat soluble colorants were 5.0 μg/kg and 10 μg/kg,respectively.The relative standard deviations of intra-assay were between 2.8% and 8.0%.The relative standard deviations of inter-assay were between 2.8% and 7.8%.The method is suitable for the determination of eight fat soluble colorants in feed.
dispersive solid phase extraction;feed;fat soluble colorants;liquid chromatography with tandem mass spectrometry(LC-MS/MS);residue
10.3969/j.issn.1004-4957.2017.04.009
2016-09-21;
2016-11-10
O657.63;TQ047.2
A
1004-4957(2017)04-0496-06
*通訊作者:吳銀良,教授級高級工程師,研究方向:農獸藥殘留分析方法研究,Tel:0574-89184044,E-mail:wupaddyfield@sina.com
