超高效液相色譜-串聯質譜法檢測花生中36種農藥及其代謝物殘留
2017-04-27 05:31:02李凌云許曉敏黃曉冬鄭姝寧劉新艷徐東輝
分析測試學報 2017年4期
李凌云,吳 華,許曉敏,林 桓,黃曉冬,鄭姝寧,劉新艷,徐東輝*
(1.中國農業科學院蔬菜花卉研究所,農業部蔬菜質量安全控制重點實驗室,農業部園藝作物生物學與種質創制重點實驗室,北京 100081;2.安捷倫科技(中國)有限公司,北京 100102)
超高效液相色譜-串聯質譜法檢測花生中36種農藥及其代謝物殘留
李凌云1,吳 華2,許曉敏1,林 桓1,黃曉冬1,鄭姝寧1,劉新艷1,徐東輝1*
(1.中國農業科學院蔬菜花卉研究所,農業部蔬菜質量安全控制重點實驗室,農業部園藝作物生物學與種質創制重點實驗室,北京 100081;2.安捷倫科技(中國)有限公司,北京 100102)
建立了花生中36種農藥及其代謝物殘留的超高效液相色譜-串聯質譜(UHPLC-MS/MS)快速檢測技術。采用乙腈提取,增強型脂質去除凈化劑(EMR-Lipid)凈化,正離子多反應監測(MRM)模式測定。結果表明,所有農藥的線性相關系數均大于0.994,在0.005,0.01,0.10 mg/kg 3個加標水平下,36種農藥的平均回收率為70.4%~119%,相對標準偏差(RSDs)為1.3%~19.4%,方法的定量下限為0.002 5~0.05 mg/kg。該方法簡便、快速,靈敏度高、凈化效果好,適用于花生中農藥多殘留的快速檢測分析。
農藥殘留;花生;脂質去除;超高效液相色譜-串聯質譜(UHPLC-MS/MS)
花生是我國重要的經濟和油料作物,在人們的日常生活中具有不可忽略的作用。由于花生在種植過程中易受多種病蟲害的侵襲,需使用大量的農藥加以防治,因此造成花生中農藥殘留超標的現象日益嚴重。花生的成分非常復雜,除了多糖、蛋白質和色素等物質外,還包含豐富的油脂[1]。這些油脂可與許多親脂性農藥互溶,嚴重干擾了目標農藥與基質的有效分離,給農藥的殘留分析帶來了很大的困難。因此,去除油脂,消除基質干擾,是建立花生基質中農藥多殘留分析技術的關鍵。
目前用于高油脂樣品分析的前處理方法主要有固相萃取[2-5]、液液萃取[6]、凝膠色譜[7-10]和基質固相分散萃取[11]等。但這些方法溶劑消耗量大,且后續仍需繁瑣的凈化步驟,耗時長,不利于農殘樣品的大批量處理和農藥事件的應急處理。近年來廣泛應用的QuEChERs方法[12-14],在處理含油脂樣品時多采用PSA+C18混合吸附劑去除油脂,但是PSA對酸類農藥有吸附,易造成這些農藥的測定結果偏低。增強型脂質去除凈化劑(Enhanced matrix removal-lipid,EMR-Lipid)是一種新型高聚物吸附劑,可以有效吸附各類脂類化合物。吳巖等[15]曾將EMR-Lipid用于玉米中三嗪類除草劑的凈化,發現EMR-Lipid能有效去除玉米基質對農藥殘留的干擾。EMR-Lipid在油脂含量更高的花生中的應用尚未見報道。
食品安全國家標準GB 2763-2014《食品中農藥最大殘留限量》規定了44種農藥在花生中的最大殘留限量,但針對這些農藥在花生中的多殘留快速檢測方法尚未建立。本研究選擇具有最大殘留限量的36 種農藥及其代謝物作為研究對象,通過使用EMR-Lipid材料改進QuEChERs凈化步驟,建立了花生中36種農藥多殘留的UHPLC-MS/MS快速檢測技術。該方法操作簡單、省時、去脂效果好、回收率高,適用于大批量花生樣品中農藥殘留的快速檢測。
1 實驗部分
1.1 儀器與試劑
LC-30A超高效液相色譜儀,LC-MS/MS-8050三重四極桿質譜儀均購自日本Shimadzu公司。
乙腈、甲醇、丙酮(HPLC級,美國JT Baker公司);乙酸銨(HPLC級,美國Fluka公司);甲酸(HPLC級,Dikma科技);水(純凈水,Milli-Q超純水儀制備);增強型脂質去除分散劑(Agilent Bond Elut QuEChERS EMR-Lipid,美國安捷倫公司);36種農藥標準品(純度>96%,德國Dr.Ehrenstorfer公司);其他試劑均為分析純,購自北京化學試劑公司。
1.2 標準溶液的配制
準確稱取10.0 mg各農藥標準品,根據農藥的溶解度選用甲醇或丙酮進行溶解,定容至10 mL,配制成1 mg/mL的標準儲備溶液。準確量取一定體積的各農藥標準儲備溶液,用甲醇定容,得到含36種農藥的混合標準工作溶液,-20 ℃保存。
1.3 樣品前處理
1.3.1 樣品制備與提取 將花生仁粉碎,過20目篩,混勻,作為試樣。精確稱取5.00 g樣品于100 mL離心管中,分別加入3 mL水和25 mL乙腈,使用勻漿機高速勻漿2 min后,加入2 g氯化鈉和2 g無水硫酸鎂,劇烈振搖1 min,5 000 r/min離心3 min,上清液待凈化。
1.3.2 樣品凈化 加入3 mL水活化EMR-Lipid除脂分散劑,混合搖勻,然后加入上清液5.00 mL,劇烈振搖1 min,5 000 r/min 離心3 min,離心后的全部溶液轉移至預先加入1 g 氯化鈉和1 g無水硫酸鎂的15 mL離心管中,劇烈振搖1 min,5 000 r/min離心3 min,上層乙腈相過0.22 μm濾膜,供UHPLC-MS/MS測定。
1.4 色譜條件
色譜柱:Waters ACQUITY UPLC BEH HSS C18(100 mm×2.1 mm,1.8 μm);流動相:A相為甲醇;B相為0.1%甲酸乙酸銨水溶液(1 mmol/L);梯度洗脫條件:0 min,20%;0~2 min,20%~60% A;2~14 min,60%~85% A;14~14.1 min,85%~95% A;14.1~17 min,95% A;17~17.1 min,95%~20% A;17.1~22 min,20% A;流速:0.3 mL/min;柱溫:40 ℃;進樣量:1 μL。
1.5 質譜條件
ESI離子源;接口電壓4 000 V;正離子掃描;MRM監測模式;霧化氣流量3 L/min,干燥氣流量10 L/min,以上2種氣體均為氮氣,加熱氣流量10 L/min,該氣體為空氣;接口溫度300 ℃;脫溶劑管溫度250 ℃;加熱模塊溫度400 ℃;碰撞氣壓力2.7×105Pa,該氣體為氬氣。
2 結果與討論
2.1 質譜條件的優化
將36種農藥分別配制成100 μg/L的標準溶液。不接色譜柱,標準溶液通過液相色譜注入質譜,進行一級質譜掃描確定各農藥的準分子離子峰。利用儀器自動優化功能,優化二級碎片離子信息,獲得定量和定性離子及質譜參數Q1(Quadrupole 1),Q3(Quadrupole 3)和碰撞能量(CE),相關參數見表1。
2.2 流動相的優化
以乙腈-水、甲醇-水、甲醇-乙酸銨水溶液和甲醇-甲酸乙酸銨水溶液分別作為流動相,考察了其對花生仁空白基質中36種農藥混合標準溶液的分析結果。結果表明,甲醇-水作流動相時,敵百蟲、高效氟吡甲禾靈等農藥的靈敏度明顯優于乙腈-水流動相。體系中加入乙酸胺,農藥的靈敏度會進一步提高。另外由于36種農藥均采用正離子掃描模式,流動相中加入少量甲酸有助于化合物電離,從而提高分析靈敏度。因此本實驗最終選擇甲醇-0.1%甲酸乙酸銨水溶液(1.0 mmol/L)作為流動相。通過優化梯度洗脫程序,36種農藥得到了最佳分離(見圖1)。
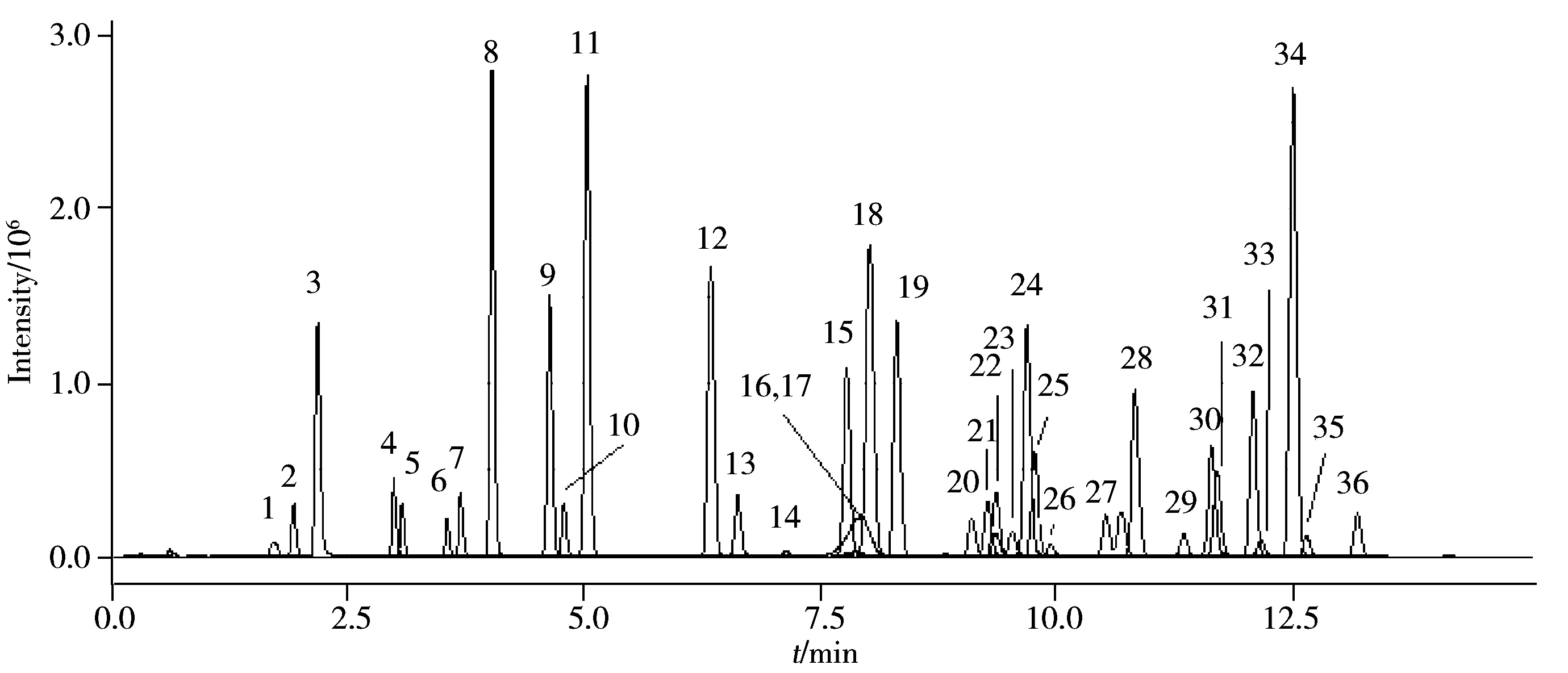
圖1 花生仁空白基質中36種農藥混合標準溶液的MRM總離子流圖(0.05 mg/kg)Fig.1 Total ion chromatogram of peanut blank matrix spiked with 36 pesticides mixed standard solution at 0.05 mg/kg in MRM mode the peak numbers denoted were the same as those in Table 1
2.3 乙腈用量的優化
比較了乙腈用量對農藥提取效率的影響。稱取5 g樣品,分別用5,10,20,25 mL乙腈進行提取,結果顯示,在36種農藥中,多菌靈的測定受乙腈用量的影響較大,5 mL乙腈提取時,多菌靈的回收率只有50%,而使用25 mL乙腈提取時,回收率則提高至82.9%。因此本實驗初步確定乙腈提取液的體積為25 mL。研究發現,僅用乙腈作為提取劑,噻吩磺隆的回收率不足70.0%,嘗試向花生中加入一定量的水,再用乙腈提取,結果發現噻吩磺隆的提取效率得到明顯改善。這可能是由于水分子占據了噻吩磺隆在花生中的吸附位點,從而使噻吩磺隆更易被提取出。本實驗繼續對花生中水的加入量進行了優化,比較了分別加入3,5,10 mL水對農藥回收率的影響。結果發現,當加水量為3 mL時,噻吩磺隆的回收率為85%,此時,其它農藥的回收率無明顯變化。但隨著加水量的進一步增大,弱極性農藥毒死蜱的回收率明顯降低。所以本實驗最終選擇加水量為3 mL。
2.4 EMR-Lipid活化水用量的優化
與傳統吸附劑不同,EMR-Lipid需要額外加入水進行活化。本實驗對活化EMR-Lipid的水量進行了優化,比較了加水量3,5,10 mL時對測定結果的影響。結果顯示,加水量對保留時間較大的化合物影響明顯大于保留時間較小的化合物,如保留時間較小的3-羥基克百威,在3種不同加水量下的回收率分別為98.3%,97.5%和98.6%,保留時間較大的毒死蜱回收率分別為85.8%,76.8%和42%。由此可見,加水量過大,會導致弱極性農藥的回收率降低,因此本實驗最終使用3 mL水活化EMR-Lipid。
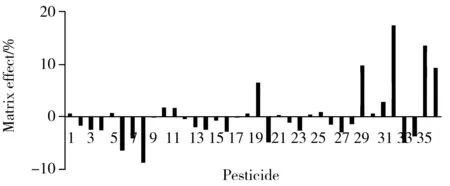
圖2 36種農藥在花生基質中的基質效應Fig.2 Matrix effects of 36 pesticides in peanut matrix the peak numbers denoted were the same as those in Table 1
2.5 基質效應考察
本文采用較為常用的相對響應值法評價基質效應,基質效應=(B/A-1)×100%[16],A為純溶劑中農藥的響應值;B為花生基質中添加相同濃度農藥的響應值。正數表示基質增強效應,負數表示基質抑制效應。研究結果見圖2,除了烯草酮和精吡氟禾草靈的基質效應分別為13.6%和17.4%以外,其它34種農藥的基質效應絕對值均在0%~10%之間,基質效應不明顯。由此可見,EMR-Lipid 能有效去除花生中的油脂,消除基質效應。
2.6 方法的線性范圍、線性關系與定量下限
采用空白的花生基質溶液,準確配制含量分別為0.50,1.0,2.5,5.0,10,25,50,100,250 μg/L的系列混合標準工作溶液。按優化實驗條件進行LC-MS/MS測定,以峰面積(y)對質量濃度(x,μg/L)作標準曲線,得到36種農藥的線性回歸方程,各農藥的線性范圍及相關系數見表1。所有農藥的相關系數(r)為0.994 2~0.999 9。其中甲基異柳磷的線性范圍為5~250 μg/L,特丁硫磷、甲草胺和乙草胺的線性范圍為1.0~250 μg/L,其余32種農藥及其代謝產物的線性范圍均為0.5~250 μg/L。
定量下限采用加標回收進行驗證,符合一定的準確度和精密度要求的最低加標濃度,確定為定量下限。結果顯示,36種農藥的LOQs為0.002 5~0.05 mg/kg(見表1),其中克百威、甲霜靈等13種農藥的LOQ為0.002 5 mg/kg,噻吩磺隆、丙線磷等22種農藥的LOQ為0.005 mg/kg,甲基異柳磷的LOQ為0.05 mg/kg。所有農藥的LOQ均低于GB 2763-2014最大殘留限量要求。
2.7 方法的回收率與精密度
采用基質匹配標準溶液-外標法定量,在花生基質中添加36種農藥及其代謝物進行加標回收率實驗,加標水平為0.005,0.01,0.1 mg/kg,每個加標水平重復6次。36種農藥在3個加標水平下的平均回收率為70.4%~119%,相對標準偏差(RSD)為1.3%~19.4%。方法的準確度和精密度均符合殘留分析的要求。

表1 36種農藥的保留時間、MRM離子對、碰撞能量、定量下限(LOQs)以及花生基質中的加標平均回收率(0.1 mg/kg)和相對標準偏差(RSDs)(n=6)Table 1 Retention times(tR),MRM transitions,collision energies,LOQs,average recoveries and RSDs of 36 pesticides in peanut spiked at 0.1 mg/kg(n=6)
(續表1)
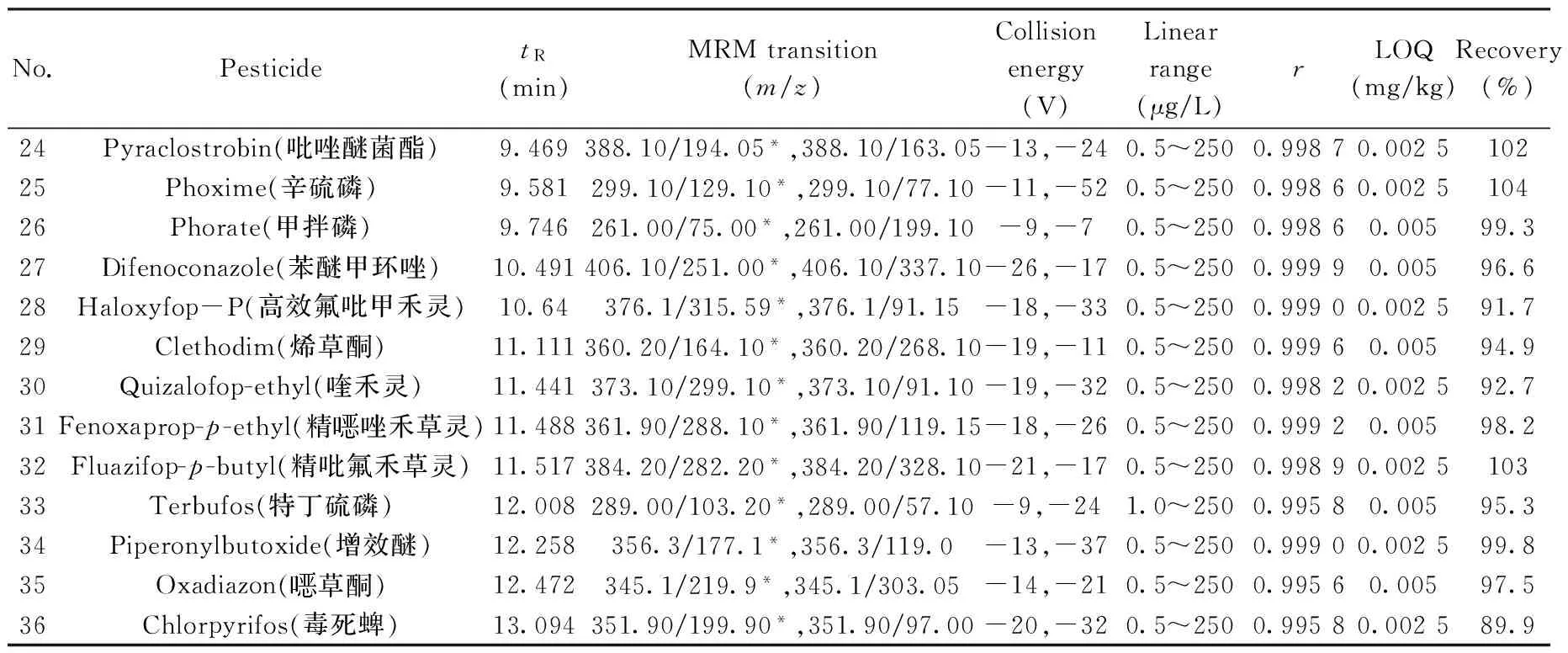
No.PesticidetR(min)MRMtransition(m/z)Collisionenergy(V)Linearrange(μg/L)rLOQ(mg/kg)Recovery(%)RSD(%)24Pyraclostrobin(吡唑醚菌酯)9.469388.10/194.05*,388.10/163.05-13,-240.5~2500.99870.00251023.425Phoxime(辛硫磷)9.581299.10/129.10*,299.10/77.10-11,-520.5~2500.99860.00251042.926Phorate(甲拌磷)9.746261.00/75.00*,261.00/199.10-9,-70.5~2500.99860.00599.35.827Difenoconazole(苯醚甲環唑)10.491406.10/251.00*,406.10/337.10-26,-170.5~2500.99990.00596.61.428Haloxyfop-P(高效氟吡甲禾靈)10.64376.1/315.59*,376.1/91.15-18,-330.5~2500.99900.002591.76.229Clethodim(烯草酮)11.111360.20/164.10*,360.20/268.10-19,-110.5~2500.99960.00594.96.330Quizalofop-ethyl(喹禾靈)11.441373.10/299.10*,373.10/91.10-19,-320.5~2500.99820.002592.75.631Fenoxaprop-p-ethyl(精唑禾草靈)11.488361.90/288.10*,361.90/119.15-18,-260.5~2500.99920.00598.23.332Fluazifop-p-butyl(精吡氟禾草靈)11.517384.20/282.20*,384.20/328.10-21,-170.5~2500.99890.00251033.333Terbufos(特丁硫磷)12.008289.00/103.20*,289.00/57.10-9,-241.0~2500.99580.00595.39.934Piperonylbutoxide(增效醚)12.258356.3/177.1*,356.3/119.0-13,-370.5~2500.99900.002599.82.435Oxadiazon(草酮)12.472345.1/219.9*,345.1/303.05-14,-210.5~2500.99560.00597.52.236Chlorpyrifos(毒死蜱)13.094351.90/199.90*,351.90/97.00-20,-320.5~2500.99580.002589.95.1
* quantitative ion
2.8 實際樣品的測定
應用所建立的方法對超市和市場上購買的7個花生樣品進行快速檢測。其中4個花生樣品被檢測出含有毒死蜱殘留(0.062,0.013,0.006 2,0.007 3 mg/kg),1個花生樣品檢出多效唑殘留(0.002 7 mg/kg),但兩種農藥的檢出量均未超出GB 2763-2014中規定的MRLs值。
3 結 論
本文采用UHPLC-MS/MS技術建立了花生中36種農藥及其代謝物殘留的定性、定量快速檢測技術。樣品經乙腈提取,EMR-Lipid除脂凈化,最后經鹽析萃取后上機分析。該方法簡便、快速、靈敏,凈化效果好,適用于花生樣品中農藥多殘留的快速檢測。
[1] Venkatachalam M,Sathe S K.J.Agric.FoodChem.,2006,54(13):4705-4714.
[2] Shimelis O,Yang Y H,Stenerson K,Kaneko T,Ye M.J.Chomatogr.A,2007,1165:18-25.
[3] Chen S A.FujianAnal.Test.(陳師安.福建分析測試),2012,21(1):8-11.
[4] Zhu Z Y,Feng M,He J,Xiong H X,Zeng Y.Chem.Anal.Meterage(朱臻怡,馮民,何健,熊華萱,曾義.分析化學計量),2010,19(1):19-21.
[5] Zheng Y B,Wang H Y.JournalofScienceofTeachersCollegeUniversity(鄭永波,王海鷹.高師理科學報),2005,25(1):39-41.
[6] Pang S,Ding X X,Li P W,Zhou H Y,Jiang J,Deng X W,Du X H.Chin.J.OilCropSci.(龐帥,丁小霞,李培武,周海燕,姜俊,鄧小偉,都曉慧.中國油料作物學報),2015,37(2):246-249.
[7] Fernández-Moreno J L,Arrebola-Liébanas F J,Garrido-Frenich A,Martinez Vidal J L.J.Chromatogr.A,2006,1111:97-105.
[8] Guardia-Rubio M,Marchal-López R M,Ayora-Caada M J,Ruiz-Medina A.J.Chromatogr.A,2007,1145:195-203.
[9] Bei F,Cui S H,Li J,Guo Q L,Lin L M.J.Instrum.Anal.(貝峰,崔淑華,李杰,郭慶龍,林黎明.分析測試學報),2012,31(Suppl):1-7.
[10] Cui S H,Guo Q L,Zhang F,Lin L M.Chin.J.Anal.Chem.(崔淑華,郭慶龍,張峰,林黎明.分析化學),2013,41(6):944-948.
[11] Zhan J,Li J D,Liu D H,Liu C,Yang G G,Zhou Z Q,Wang P.FoodChem.,2016,194:319-324.
[12] Chen X,Cheng L,Qu S C,Huang D L,Liu J C,Cui H,Jia Y B,Ji M S.Chin.J.Chromatogr.(陳溪,程磊,曲世超,黃大亮,劉佳成,崔晗,賈彥波,紀明山.色譜),2015,33(10):1080-1089.
[13] Lehotay S J,Mastovska K,Yun S J.J.AOACInt.,2005,88(2):630-638.
[14] Cunha S C,Lehotay S J,Mastovska K,Fernandes J O,Beatriz M,Oliveira P P.J.Sep.Sci.,2007,30:620-632.[15] Wu Y,Zhao W,Liu Y,Jiang B,Wei D X,Gou Y,Li L L,Han F,Zu Y G.Chin.J.Anal.Chem.(吳巖,趙偉,劉永,姜冰,魏東旭,勾越,李麗麗,韓峰,祖元剛.分析化學),2016,4(6):950-957.
[16] Chambers E,Wagrowski-Diehl D M,Lu Z L,Mazzeo J R.J.Chromatogr.B,2007,852(1/2):22-34.
Rapid Detection of 36 Pesticide Residues in Peanut by Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry
LI Ling-yun1,WU Hua2,XU Xiao-min1,LIN Huan1,HUANG Xiao-dong1,ZHENG Shu-ning1,LIU Xin-yan1,XU Dong-hui1*
(1.Key Laboratory of Biology and Genetic Improvement of Horticultural Crops,Ministry of Agriculture,Key Laboratory of Quality & Safety Control for Vegetable Products,Ministry of Agriculture,Institute of Vegetables and Flowers,Chinese Academy of Agricultural Sciences,Beijing 100081,China;2.Agilent Technologies,Beijing 100102,China)
A multiresidue analytical method was developed for the rapid detection of 36 pesticides in peanut using ultra high performance liquid chromatography-tandem mass spectrometric technique(UHPLC-MS/MS).Peanut samples were extracted with acetonitrile,and then cleaned up with QuEChERS EMR-Lipid.The extract was detected by UHPLC-MS/MS.The positive ion mode and multiple reaction monitoring(MRM) mode were used to identify and quantify 36 pesticide residues in peanut.All pesticides had good linearity with correlation coefficients above 0.994.The average recoveries of the 36 pesticides ranged from 70.4% to 119% with relative standard deviations(RSDs) of 1.3%-19.4% at spiked levels of 0.005,0.01,0.10 mg/kg.The quantitation limits of this method were in the range of 0.002 5-0.05 mg/kg. With the advantages of simplicity,rapidness,sensitivity and good purifying effect,the method was suitable for the rapid determination of pesticide residues in peanut.
pesticide residues;peanut;enhanced matrix removal;ultra high performance liquid chromatography-tandem mass spectrometry(UHPLC-MS/MS)
10.3969/j.issn.1004-4957.2017.04.010
2016-10-24;
2016-11-30
國家蔬菜產品質量安全風險評估項目(GJFP2017002);國家重點研發計劃項目(2016YFD0200200)
O657.63;F767.2
A
1004-4957(2017)04-0502-05
*通訊作者:徐東輝,研究員,研究方向:農產品質量安全,Tel:010-82106963,E-mail:xudonghui@caas.cn
