美國和歐盟的罕用藥研發激勵政策對比研究與啟示Δ
2017-07-03 14:57:00田麗娟沈陽藥科大學工商管理學院沈陽110016
中國藥房 2017年16期
楊 莉,田麗娟,林 琳(沈陽藥科大學工商管理學院,沈陽 110016)
·藥事管理·
美國和歐盟的罕用藥研發激勵政策對比研究與啟示Δ
楊 莉*,田麗娟,林 琳(沈陽藥科大學工商管理學院,沈陽 110016)
目的:為構建和完善我國的罕用藥研發激勵政策提供參考和建議。方法:從罕用藥的立法沿革、研發罕用藥的激勵措施與效果方面對美國和歐盟的罕用藥研發激勵政策進行對比,并為我國完善相關政策提供建議。結果與結論:美國與歐盟的罕用藥激勵政策分別始于1983年美國《罕用藥法案》與1999年歐盟《罕用藥管理規范》,之后通過不斷完善,形成了較為完備的體系。美國與歐盟在罕用藥的認定標準、認定程序、具體激勵措施(研發資助、稅收減免、費用減免、微型與中小企業額外激勵、市場獨占、特殊審批程序)等方面有所差異,如在費用減免方面,美國對處方申請費用、生產費用和藥物確認費用進行減免,而歐盟對協議幫助費用、初始和后續要求費用,審批前的檢查費用和首次上市申請費用依類型按一定比例進行減免。罕用藥激勵政策推行后,其資格認定數量及上市數量大幅增加、微型與中小型企業成為罕用藥研發的生力軍、研發投資涵蓋各類疾病治療領域、罕用藥研發成為藥物創新和生物技術發展的主要方向。我國應該盡快確定罕用藥研發激勵的相關立法、設立罕用藥的資格認定、從多方面入手完善罕用藥研發激勵具體措施,同時加強與其他國家在罕用藥資格認定和研發激勵方面的合作。
罕用藥;研發激勵;美國;歐盟;對比;研究
罕用藥由于臨床研究開展困難、研發成本難以收回等因素成為許多醫藥企業不愿意涉足的領域。目前,世界上超過35個國家都推行并實施了不同程度的罕用藥研發激勵政策[1]。美國和歐盟作為世界上對罕用藥研發激勵最為傾斜的兩個國家(地區),是許多學者研究的對象,但是針對這兩個國家(地區)的罕用藥研發激勵政策的對比研究較少。因此,在本研究中,筆者從罕用藥的認定、立法、具體措施以及實施效果等多方面對美國和歐盟的罕用藥研發激勵政策進行對比研究,以期為我國罕用藥研發激勵政策的構建和完善提供建議。
1 美國和歐盟罕用藥研發激勵的立法沿革
1983年1月4日,美國頒布并實施了《罕用藥法案》,開啟了通過立法對罕用藥進行研發激勵之門。1999年,歐盟委員會頒布了《罕用藥管理法規》,鼓勵歐盟的醫藥企業進行罕用藥的研發,該法于2000年4月28日正式實施。美國和歐盟罕用藥研發激勵立法比較見圖1。
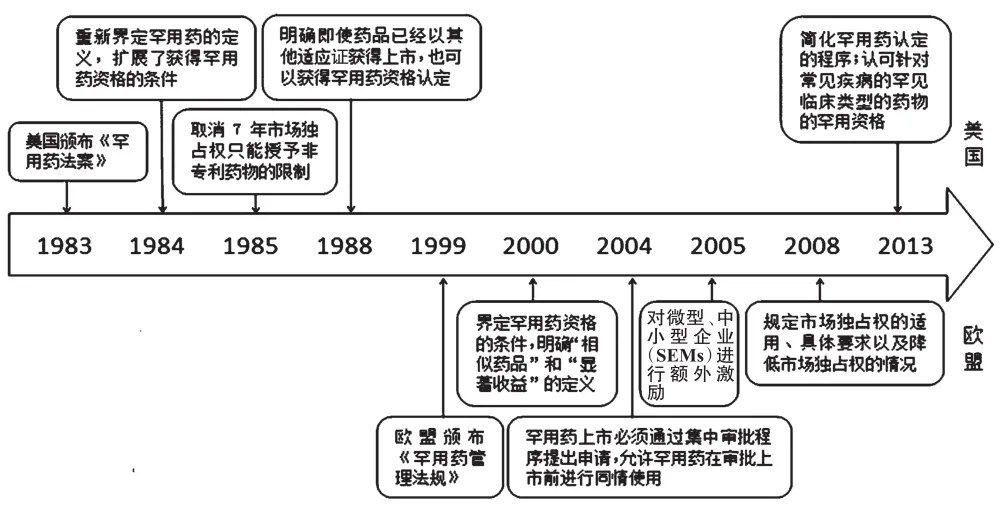
圖1 美國和歐盟罕用藥研發激勵立法比較Fig 1 Comparisons of the R&D incentive legislations for orphan drugs in USA and the EU
由圖1可知,美國對罕用藥資格認定的范圍不斷放寬,研發激勵措施也逐漸加強。1992年和1994年曾有兩次限制罕用藥市場獨占權的修訂提案,而國會最終基于“藥雖貴但強于無藥可醫”的保守考慮未通過此項修訂案,這也反映了美國對罕用藥研發激勵的強烈傾斜態度[2]。除了《罕用藥法案》之外,美國還于1992年頒布了《罕用藥法實施辦法》,對罕用藥的研發激勵和管理進行了更為詳細的規定;2002年通過了《罕見疾病藥物發展法案2002》,對《罕用藥法案》的內容進行了補充。
歐盟繼《罕用藥管理法規》之后多次頒布新的法規繼續補充和完善罕用藥研發激勵的內容。相比于美國,歐盟對罕用藥資格的限定更多。此外,歐盟藥品管理局(EMA)還出臺了一系列指南,對罕用藥研發享受到的一系列優惠政策,如費用減免、協議幫助等加以說明。
2 美國和歐盟罕用藥研發激勵的具體措施比較
2.1 罕用藥資格的認定標準
美國罕用藥資格的獲得需要滿足以下條件:治療疾病的患病人數不滿20萬,或者患病人數超過20萬,但合理預期的銷售收入無法收回研發成本。2013年8月實施的《罕用藥法案》修正案,將常見疾病的罕見臨床類型的治療藥物也納入可獲得罕用藥資格的范圍。
歐盟罕用藥資格只授予用來預防、診斷和治療危及生命、慢性消耗性疾病的藥品,并且該疾病在歐洲患病人數不超過萬分之五,或者如果沒有激勵措施,該藥品上市后的收入無法充分地回收其研發成本。除此之外,還應該滿足以下條件:除了該藥品,目前還沒有令人滿意的預防、診斷和治療該疾病的藥品或方法,或者相比于現存的藥品或方法,該藥品對患者有顯著收益(具有臨床優勢或對患者有顯著的貢獻,如更好的治療指征、不同的作用機制、更高的安全性、更方便的給藥途徑以及更便宜的價格等)[3]。
典型國家或地區的罕用藥資格認定標準見表1。
由表1可見,美國執行了最寬松的標準,而歐盟則執行了最嚴格的標準。

表1 典型國家或地區的罕用藥資格認定標準Tab 1 Orphan drug designation standards in typical countriesor regions
2.2 罕用藥資格的認定程序
美國罕用藥資格的認定部門是FDA的罕用藥開發辦公室(OOPD)。歐盟的罕用藥資格由EMA設立的罕用藥委員會(COMP)負責審評,并出具評估意見,但由歐盟委員會(EC)最終決定是否授予罕用藥資格。美國和歐盟的罕用藥資格認定程序見圖2。
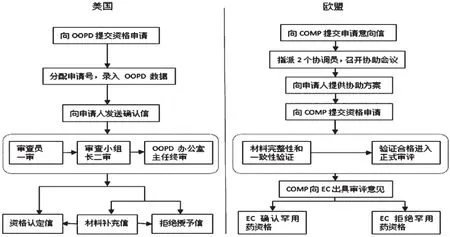
圖2 美國和歐盟的罕用藥資格認定程序Fig 2 O rphan d rug designation procedure in USA and the EU
由圖2可見,歐盟對罕用藥資格的認定程序更為復雜。除此之外,在審評的具體細節上,美國和歐盟也有較多區別,詳見表2。

表2 美國和歐盟的罕用藥資格認定具體內容比較Tab 2 Comparison of the specific contents of orphan drug designation in USA and the EU
由于歐盟在罕用藥的認定上有嚴格的時限要求,因此從提交正式申請算起,整個罕用藥資格的認定為120~180天。而美國對時限無明確要求,因此整個罕用藥的認定為30~365天,其中有75%的認定都是在90天內完成的。隨著近年來申請數目的增加,FDA罕用藥認定時限的目標是將75%的申請控制在120天內完成審核。截至2015年12月31日,FDA共收到5 561份申請,其中有3 612份申請獲得了罕用藥資格認定,通過率為65%[4];歐盟共收到2 385份申請,得到罕用藥資格的申請為1 596份,通過率為67%[5]。因此,從認定的效率和通過率來說美國和歐盟的差別不大。雖然美國在提交申請前沒有專門的協助會議,但是在申請過程中FDA也會給申請人提供一些建議。
2.3 激勵措施
美國和歐盟的罕用藥研發激勵措施比較見表3。

表3 美國和歐盟的罕用藥研發激勵措施比較Tab 3 Com parison of the orphan drug incentivemeasures in USA and the EU
2.3.1 研發資助 美國的罕用藥研發資助:一是美國國立衛生研究院(NIH)提供的項目資助,傾向于罕用藥研發的基礎研究;二是一些公司合作伙伴組織(PPPs)提供的針對一些特定疾病的研究的資助,例如國際艾滋病疫苗倡議組織(IAVI);三是OOPD辦公室的罕用藥研發項目,主要資助的是罕用藥的臨床研究,該項目1983年起實施以來,迄今已經收到1 800多份申請,有600余項項目得到了資助,資助的項目中已經有45個罕用藥獲得了上市批準[6]。
EMA不直接對罕用藥的研發提供資助,由EC通過框架計劃對罕用藥的研發提供資金支持。《第七框架計劃(2007-2013)》(FP7)從2009年起將獲得罕用藥資格作為獲得資助的必要條件。FP7共投資6.2億歐元資助超過120個罕用藥研發項目,資助范圍包括罕用藥的基礎研究、臨床前研究和臨床研究,涵蓋各類罕見疾病。2014年,新的研究與創新框架計劃——“地平線2020”(Horizon 2020)正式啟動,為期7年(2014-2020年)。2014-2015年,“地平線2020”已經資助了腫瘤、呼吸、消化等方面共20余個罕用藥研發。
2.3.2 稅收減免 美國的罕用藥稅收減免用于臨床試驗階段的支出,無論該罕用藥最終能否獲得上市許可都可享受減免。美國國會的技術評估辦公室(OTA)的研究表明,稅收減免可以將臨床研究成本至少降低24%,僅在Ⅲ期臨床階段,罕用藥的研發成本就是非罕用藥的1/2[7]。由于臨床試驗是整個藥物研發過程中耗資最大的階段,因此稅收減免帶來的收益非常可觀,也成為很多醫藥企業是否繼續開展臨床研究的重要考量因素。
考慮到享受稅收減免的罕用藥最終不一定能夠獲得上市,資金不一定能得到最佳使用,故歐盟并未制定稅收減免政策,而是通過設立更長的市場獨占期來代替稅收減免。
2.3.3 費用減免 美國的罕用藥費用減免分為處方申請費用、生產費用和藥物確認費用。如果獲得罕用藥資格,處方藥申請費是自動豁免的。生產費用和藥物確認費的減免則需要提交申請,減免的比例不固定。
歐盟的費用減免需要提出申請,包括協議幫助費用、初始和后續要求費用,審批前的檢查費用和首次上市申請費用,減免的比例是固定的。
2006-2015年美國和歐盟的罕用藥的費用減免總額均呈現逐年上漲的趨勢,詳見表4(數據來源于醫藥市場研究機構Evaluate Pharma)。
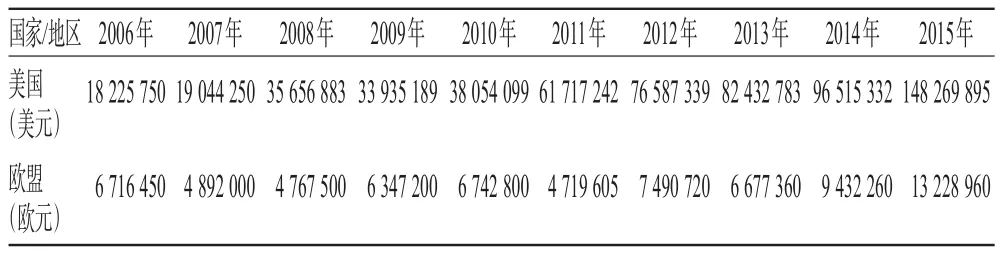
表4 2006-2015年美國和歐盟罕用藥費用減免總額Tab 4 Com parison of fee reduction of orphan drugs in USA and the EU during 2006-2015
2.3.4 協議幫助 美國和歐盟的協議幫助主要包括罕用藥資格的保持和罕用藥的批準上市。協議幫助的方式是由申請人向OOPD或人用藥品委員會(CHMP)提出申請,并列出相關問題,由OOPD和CHMP給予解答以解決罕用藥在研發過程中有關問題,降低因研究設計不當而導致的取消罕用藥資格和未通過上市批準的風險。申請人可以申請協議幫助的次數沒有限制。協議幫助在幫助罕用藥上市方面發揮了積極的作用,截至2015年底,CHMP共完成了951份協議幫助,被批準上市的罕用藥中,超過50%都接受過協議幫助。
2.3.5 SEMs額外激勵 歐盟從2005年(罕用藥的申請必須通過集中審批程序批準)后開始對SEM s施行額外的罕用藥研發激勵政策。企業需要向EMA提交申請,由EMA審核確定其滿足SEMs的標準,才能享受此激勵。截至2015年底,歐盟已批準上市的罕用藥中,有25個來自SEMs。
2.3.6 市場獨占 美國和歐盟分別給予罕用藥7年、10年的市場獨占期。如果該罕用適應證可以用于兒科,并進行了兒科臨床研究,則獨占期可以分別延長6個月、2年。同時,美國和歐盟也規定了喪失罕用藥資格的情況:某藥品比已上市的藥品更具有臨床優越性(更有效、更安全或對患者的健康更有益);或獲得罕用藥獨占的藥品供應不足,無法滿足需求;或獲得罕用藥獨占權的藥品持有人同意。
此外,歐盟還規定,當罕用藥5年獨占期結束,如果有成員國通知EMA該藥品已經不符合罕用藥資格的標準,則該罕用藥的市場獨占期將會被降為6年。
2.3.7 特殊審批 美國和歐盟的罕用藥特殊審批程序比較見表5。

表5 美國和歐盟的罕用藥特殊審批程序比較Tab 5 Com parison of special approval procedure of orphan drugs in USA and the EU
從表5可以看出,美國的加速審評和優先審評與歐盟的條件審評和加速審評類似,而其他的審評程序美國和歐盟各有特色。申請人想要通過特殊審評程序獲得上市批準必須向FDA或EMA提出申請。截至2015年9月底,FDA批準罕用藥所需的平均時間為315天,非罕用藥為378天;EMA批準罕用藥所需的平均時間為210天,非罕用藥為270天[8]。可見,罕用藥上市速度明顯要快于非罕用藥。
2.4 美國和歐盟在罕用藥研發激勵方面的合作
2.4.1 罕用藥資格申請 2007年,FDA和EMA合作引進了罕用藥資格通用申請格式,激勵申請人在美國和歐盟并行提交申請。通用格式既包含了FDA和EMA的共同信息要求,也包含各機構的單獨要求,申請人可以以通用格式同時在美國和歐盟提出申請。通用申請格式的引進大大降低了申請人的行政成本,提高了申請效率。迄今為止,EMA的罕用藥資格申請有50%左右是并行提交到FDA的,有40%左右使用了通用申請格式。
2.4.2 年度報告 獲得罕用藥資格后,FDA要求14個月內提交年度發展報告,此后每年提交1次直至獲得上市批準。EMA也有提交年度發展報告的要求。2011年開始,美國和歐盟宣布接收通用模板的年度報告。年度報告的內容主要包括研究計劃、已完成的研究工作、遇到的困難以及潛在的風險等。依據研究報告的內容,FDA和EMA有權撤銷罕用藥資格。
2.4.3 協議幫助 FDA和EMA建立了并行的協議幫助程序。當申請人獲得罕用藥資格后,可以同時向FDA和EMA申請協議幫助。同時FDA和EMA也可通過協商,對申請人同時開展協議幫助。
FDA和EMA的合作,也促進了很多醫藥企業積極申請在美國和歐盟同時獲得認定和上市。截止到2015年底,歐盟獲得罕用藥資格的藥品中有53%同時在美國獲得了罕用藥資格,獲得上市的罕用藥中有49%同時在美國獲得了上市[9]。
3 美國和歐盟的罕用藥研發激勵效果比較
推出罕用藥激勵政策之后,美國和歐盟的罕用藥資格認定情況和批準上市情況見圖3(數據來源于FDA和EMA網站[4-5]),效果比較見表6(數據來源于Drug Discovery Today,其中V類別是解剖學治療學及化學分類系統代碼中的規定)。
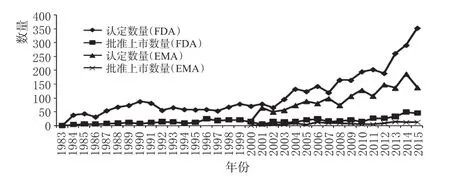
圖3 美國和歐盟的罕用藥資格認定和批準上市情況Fig 3 Orphan drug designation and approval for listing in USA and the EU
3.1 罕用藥資格認定數量及上市數量大幅增加
從圖3可以看出,美國和歐盟的罕用藥資格的認定數量均呈現逐年上漲趨勢,且每年都有一定數量的罕用藥獲得上市,上市的罕用藥總量呈現平穩增長。
從取得上市資格的罕用藥占獲得罕用藥資格的藥品的比例來看,美國為15.1%,歐盟為7.6%。雖然歐盟的罕用藥激勵政策比美國滯后17年左右,但是資格認定的增長速度很快。但是資格認定中獲得上市批準的比例卻比美國低很多。如果以罕用藥上市的數量和比例作為衡量激勵政策作用的重要標準,則美國的激勵政策效果更為顯著。
3.2 中小型企業成為罕用藥研發的生力軍
由表6可見,在申請類型上美國和歐盟極為相似。提出罕用藥資格申請最多的公司集中在全球排名100以后的制藥公司。一些中小企業,特別是一些生物科技公司看準了罕用藥研發激勵政策(特別是市場獨占政策)提供的資金支持和研發幫助,以罕用藥作為目標市場,在此基礎上逐漸發展壯大,比如美國的BioMarin、Ae-gerion制藥公司,歐盟的Actelion制藥公司。
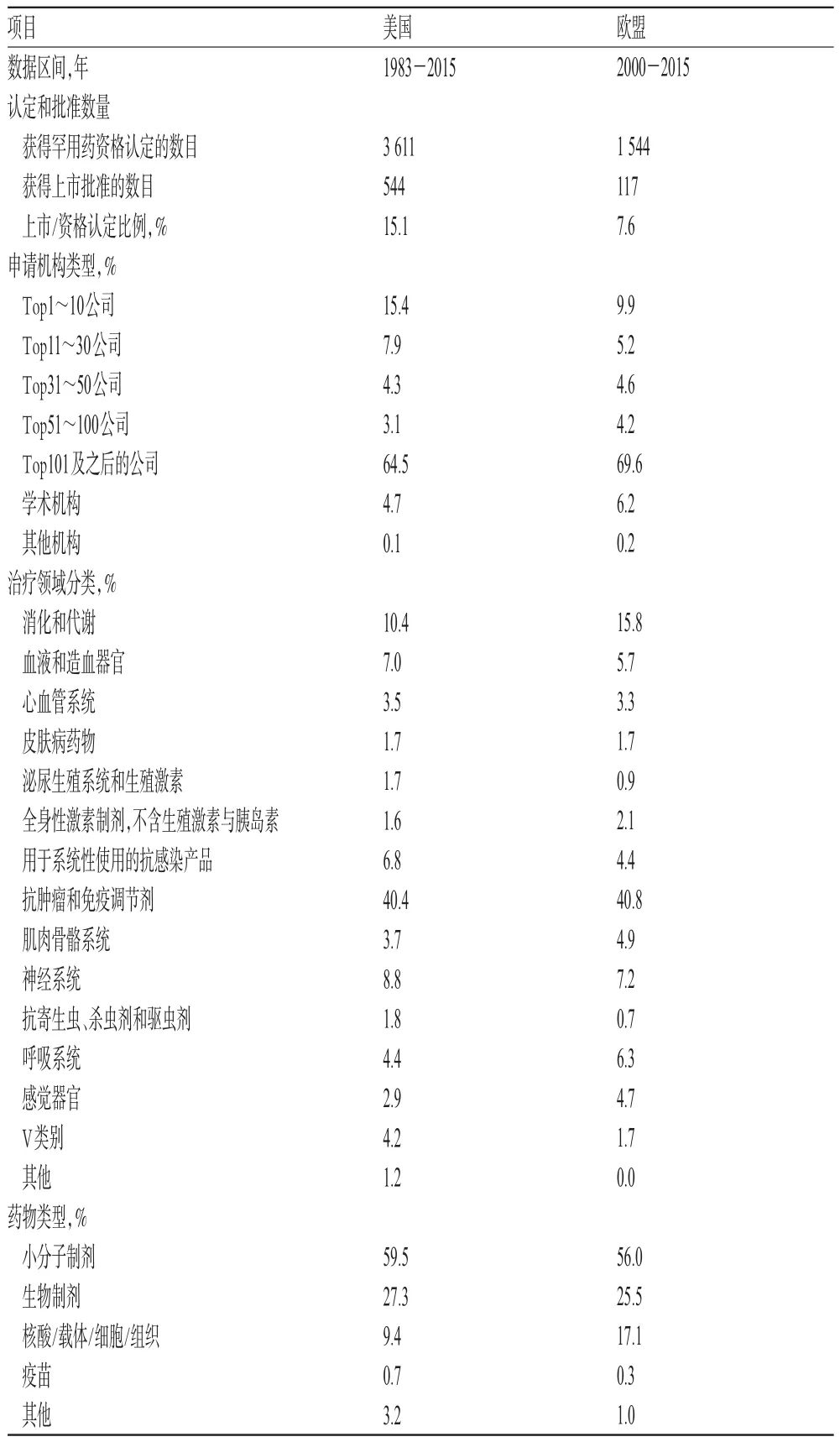
表6 美國和歐盟的罕用藥激勵政策的效果比較Tab 6 Comparison of incentive policy effects of orphan d rugs in USA and the EU
3.3 罕用藥研發投資涵蓋各類疾病領域
由表6可見,美國和歐盟申請數量排第1位的都是在研發投資上更容易回收的抗腫瘤和免疫調節藥物。同時,在其他治療領域上,美國和歐盟的罕用藥研發資本均有涉足。且在治療領域分類的申請排序上,美國和歐盟的差別也不大。
3.4 罕用藥研發已經成為藥物創新和生物技術發展的主要方向
由表6可見,美國和歐盟的藥物類型,排名第1位的均為小分子制劑,第2位為生物制劑。近年來,隨著研發突破越來越困難,研發投資越來越多,很多制藥公司將研發戰略從常見病藥物轉移到罕用藥的研發上,生物醫藥產業成為罕用藥研發的主力。因此,美國近些年來批準上市的罕用藥物占每年新化學藥物的1/3左右,占每年新生物藥物的2/3左右[10]。
4 美國和歐盟罕用藥研發激勵政策對我國的啟示
我國目前尚未建立類似于美國和歐盟的系統的罕用藥研發激勵政策。主要的有關罕用藥研發激勵的政策包括:《藥品注冊管理辦法》第45條規定對罕見病藥物可以進行特殊審批;《新藥注冊特殊審批管理規定》中提出,在必要情況下,罕見病藥物可以提出減少臨床受試人群或豁免臨床試驗的申請;《關于藥品注冊審評審批若干政策的公告》中提出對罕見病等疾病的創新藥注冊申請實行單獨排隊,加快審評審批。因此,目前我國罕用藥的研發激勵政策主要集中在特殊審評,與美國和歐盟的相關政策存在較大差距。
4.1 確定罕用藥研發激勵的相關立法
美國和歐盟罕用藥研發激勵的立法層級較高,都屬于立法委員會通過的法律。我國目前最高層級的與藥品相關的專門立法是《中華人民共和國藥品管理法》,但無專門的罕用藥研發激勵相關條款。涉及到罕用藥研發激勵的上述條款屬于部門規章或補充和說明,條款比較簡單,缺乏詳細具體的內容。因此,我國首先需要解決的是罕用藥研發激勵的立法問題。筆者建議我國在《藥品管理法》中增加藥品研發激勵的相關條款,例如鼓勵和促進罕用藥的研發,并由國家食品藥品監督管理總局出臺詳細的政策解釋,完善《藥品注冊管理辦法》中關于罕用藥注冊的相關條款。
4.2 設立罕用藥的資格認定
目前,我國的相關法規和文件中出現的都是“罕見病藥物”的表述,強調的是藥物所治療疾病的患病率,但是即使對罕見病也沒有作確切定義。因此,我國罕用藥的資格認定首先應該明確罕見疾病的定義。在罕用藥資格判定方面,除日本以外,其他國家都將無法收回投資作為可以獲得罕用藥資格的條件之一。很多創新藥物都會面臨上市后多年無法收回成本的尷尬境地,因此我國可以考慮將這類藥物納入罕用藥的范圍;同時,應借鑒歐盟設立限制條件,例如加入針對一定危重程度的疾病、具有臨床優越性等要求。此外,研究項目的可行性通過申請研發資助時加以審評,筆者認為并非必要條件。是否被其他管理當局拒絕授予罕用藥資格僅作為參考條件,因為每個國家和地區的罕見疾病并不相同。
筆者建議我國成立專門的罕用藥資格認定機構,對罕用藥資格進行審評;確定罕用藥資格的認定程序,并將罕用藥資格認定程序作為罕用藥注冊批準的前置程序;由于我國目前的藥品行政審批都有具體的時限要求,因此對罕用藥資格認定也要設立具體的時限;此外,相關部門應做好信息公開工作,及時在相關網站上對獲得罕用藥資格的藥品進行公開,并接受社會監督;年度發展報告應該作為維持或撤銷罕用藥資格的必要條件。
4.3 完善罕用藥研發激勵具體措施
4.3.1 研發資助 我國每年投入大量的資金對新藥研發進行資助,例如重大新藥創制、國家重點新產品計劃等。對罕用藥研發提供資金支持是研發激勵最直接的措施,建議我國設立罕用藥研發的專項資助,做到臨床與基礎研究并重。相關部門可以針對特定疾病設立專項,引導資本投入到市場不愿涉足的疾病領域中去。此外,獲得研發資助也需要經過申請和篩選,使資金切實運用到具有可行性的項目上。
4.3.2 費用減免 相比于美國和歐盟,我國目前還未推行處方藥申報付費制度,因此費用減免在當前意義不大,但是可以免除協議幫助費用。
4.3.3 稅收減免 關于稅收減免,筆者建議采用歐盟的形式,可以不制定。第一由于稅收減免不是靠國家食品藥品監督管理總局一個部門就可以決定的,這涉及到我國的稅務部門,會增加政策制定的難度;第二,獲得罕用藥資格的藥品不一定能夠獲得上市,如果沒有獲得上市,稅收減免的后果則是由政府來承擔的。
4.3.4 協議幫助 我國《藥品注冊特殊審批管理規定》中明確提出,已獲準實行特殊審批的注冊申請,原國家食品藥品監督管理局藥品審評中心應建立與申請人溝通交流的工作機制,共同討論相關技術問題;申請人在完成某一階段臨床試驗及總結評估后,可就下列問題向原國家食品藥品監督管理局藥品審評中心提出溝通交流申請:重大安全性問題、臨床試驗方案和階段性臨床試驗結果的總結與評價。這其實已經是協議幫助的一部分了。我國可以借鑒美國和歐盟的經驗,在獲得罕用藥資格前,或者提交注冊前就建立起與國家食品藥品監督管理總局的溝通,對罕用藥資格的認定和上市都能提供科學的建議和幫助。
4.3.5 SEMs額外激勵 我國SEMs普遍存在創新能力薄弱和研發資金不足的情況。建議在研發資助、協議幫助方面對SEMs進行一些傾斜。
4.3.6 市場獨占 給予罕用藥一定期限的市場獨占是罕用藥研發激勵的核心措施,也是最強有力的措施。前文所述的措施都是在罕用藥上市之前發揮作用的,而市場獨占則是罕用藥上市之后獲得的一定時期的市場壟斷,直接決定了罕用藥上市之后的利潤和預期回報,也是醫藥企業最重視的。市場獨占首先需要確定的是獨占期,要基于罕用藥研發成本和預期收益設定合理的期限。其次,建議參考歐盟的經驗,對獨占期進行靈活處理,例如當該藥品已經不符合罕用藥的標準,或者銷售收入超過某一標準后取消或縮短獨占期。對于全新的罕用藥和“老藥新用”的罕用藥在獨占期的設定上也可以區別對待。最后,還要規定打破市場獨占的情況,從臨床優越性、是否征求了獨占人的許可和市場供求角度來考慮。我國的新藥監測期制度,規定獲得生產許可之日起2年之內如果不生產,則取消監測期。為了和此制度接軌,也可以設立2年不生產則取消市場獨占的規定。4.3.7 特殊審批 我國雖然已經建立了罕用藥上市的特殊審批制度,但是具體程序還有待改善:一是設置多樣化的審批程序,根據不同情況設立不同的特殊審批程序;二是明確特殊審批需要滿足的技術標準和要點,特別是上市終點指標;三是明確特殊審批的藥品上市后的風險效益評價工作。同美國和歐盟一樣,特殊審批程序應該是我國藥品注冊改革的一項重要內容,不僅適用于罕用藥。
4.4 加強與其他國家的合作
目前,在罕用藥的資格認定和研發激勵方面,美國與歐盟、日本已經建立了合作關系。我國雖然短期之內不太可能在資格申請、協議幫助、年度報告等方面和這些國家或地區馬上建立合作和聯系,但是可以做好國際合作的準備,以利于促進我國醫藥企業占據國際罕用藥市場。
綜上所述,我國應該盡快確定罕用藥研發激勵的相關立法、設立罕用藥的資格認定、從多方面入手完善罕用藥研發激勵具體措施,同時加強與其他國家在罕用藥資格認定和研發激勵方面的合作。
[1]Gamm ie T,Lu CY,Babar ZU.Access to orphan drugs:a comprehensive review of legislations,regulationsand policies in 35 countries[J].PLoSOne,2015,10(10):e0140002.
[2]Braun MM,Farag-El-Massah S,Xu K,etal.Emergency of orphan drugs in the United States:a quantitative assessmentof the first25 years[J].NatRev Drug Discov,2010,9(7):519-522.
[3] Ogbah R,Associate L,Pharmalink G.Orphanmedicinal products:a European process overview[J].Regulatory Rapporteur,2015,12(2):5-11.
[4] FDA.Search orphan drug designations and approvals [EB/OL].(2016-06-25)[2016-07-15].http://www.accessdata.fda.gov/scripts/opdlisting/oopd/index.cfm.
[5] EMA.Orphan medicines figures:2000-2015[EB/OL].(2016-03-03)[2016-07-06].http://www.ema.europa. eu/docs/en_GB/document_library/Other/2015/04/ WC500185766.pdf.
[6] FDA.Orphan products grants program[EB/OL].(2016-01-13)[2016-07-16].http://www.fda.gov/ForIndustry/ DevelopingProductsforRareDiseasesConditions/Whom to-ContactaboutOrphanProductDevelopment/default.htm.
[7] Fellows GK,Hollis A.Funding innovation for treatment for rare diseases:adopting a cost-based yardstick approach [J].Orphanet JRare Dis,2013,8(1):180-185.
[8] Evaluate Pharma.Orphan drug report 2015[EB/OL].(2016-01-30)[2016-07-05].http://info.evaluategroup. com/rs/607-YGS-364/images/EPOD15.pdf.
[9] MurakamiM.Matched analysis on orphan drug designations and approvals:cross regional analysis in the United States,the European Union,and Japan[J].Drug Discov Today,2016,21(4):544-550.
[10] Galati F,BigliardiB.The unintended effectof the Orphan Drug Act on the adoption of open innovation[J].Sci Public Policy,2016,doi:10.1093/scipol/scw001.
Com parative Study and Enlightenment of the R&D Incentive Policies for Orphan Drugs in USA and the EU
YANG Li,TIAN Lijuan,LIN Lin(College of Business Adm inistration,Shenyang Pharmaceutical University,Shenyang 110016,China)
OBJECTIVE:To provide references and suggestions for building and improving the R&D incentive policies for orphan drugs in China.METHODS:The R&D incentive policies for orphan drugs in USA and the EU were compared in aspects of its legislative history,incentivemeasures and effects.And suggestions for improving related policies in China were put forward.RESULTS&CONCLUSIONS:The R&D incentive policies for orphan drugs in USA and the EU respectively started from Orphan Drug Act in USA(1983)and Orphan Drug Management Specification in the EU(1999),then formed relatively complete system by continuous improvement.The USA and the EU showed differences in its certification standard,procedure and specific incentives [R&D funding,tax deduction,fee reduction,additional incentives for micro and small and medium enterprises(SMEs),market exclusivity and special approval procedure],etc.In terms of fee reduction,for example,prescription application fee,production cost and drug confirmation fee were exempted in USA,while arrangement assist fee,initial and follow-up fee,checking fee before approval,initial listing type were reduced to a certain percentage in the EU.After developing incentive policies for orphan drugs,there is great increase in numbers of recognized qualifications and listing,SMEs have become new force in orphan drug R&D,R&D investment covers all types of diseases,orphan drug R&D are becoming themain direction of drug innovation and biotechnology development.China should determ ine the relevant legislation of R&D incentives for orphan drugs as soon as possible,set certification and improve specific measures of R&D incentives for orphan drugs from multiple aspects,while strengthen the cooperation w ith other countries in qualification and R&D incentives.
Orphan drug;Research and development incentive;USA;EU;Comparative;Research
R95
A
1001-0408(2017)16-2161-06
2016-10-12
2017-04-06)
(編輯:劉明偉)
國家社會科學基金項目(No.13CFX086);遼寧省教育廳科學研究一般項目(No.W 2014119);沈陽藥科大學中青年教師事業發展支持計劃
*副教授,博士。研究方向:藥事法規與藥物政策。E-mail:yanglishanxi@126.com
DOI10.6039/j.issn.1001-0408.2017.16.01
猜你喜歡
車主之友(2022年6期)2023-01-30 08:01:04
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
車主之友(2022年4期)2022-11-25 07:27:30
車主之友(2022年5期)2022-11-23 07:24:48
車主之友(2022年4期)2022-08-27 00:57:48
車主之友(2022年5期)2022-04-06 11:54:26
小學生優秀作文(低年級)(2018年6期)2018-05-19 01:54:28
中國衛生(2016年5期)2016-11-12 13:25:28
中國衛生(2015年9期)2015-11-10 03:11:14
中國衛生(2015年5期)2015-11-08 12:09:48
