同步輻射高壓單晶衍射實驗技術?
2017-07-31 05:59:38李曉東李暉李鵬善
物理學報 2017年3期
李曉東李暉李鵬善
1)(中國科學院高能物理研究所多學科中心,北京 100049)2)(北京工業大學固體微結構與性能研究所,北京 100124)(2017年1月10日收到;2017年1月13日收到修改稿)
專題:高壓下物質的新結構與新性質研究進展
同步輻射高壓單晶衍射實驗技術?
李曉東1)?李暉2)李鵬善1)
1)(中國科學院高能物理研究所多學科中心,北京 100049)2)(北京工業大學固體微結構與性能研究所,北京 100124)(2017年1月10日收到;2017年1月13日收到修改稿)
利用同步輻射光源開展高壓X射線衍射(X-ray diff raction,XRD)研究已有近四十年的歷史,并且已經取得了非常豐碩的成果.單晶XRD作為高壓研究的一部分,在同步輻射裝置上的應用也有了接近三十年的歷史.近年來,隨著同步輻射光學技術以及高壓技術、特別是大衍射窗口金剛石對頂砧壓腔(diamond anvil cell,DAC)的改進與發展,同步輻射高壓單晶衍射實驗方法在高壓研究中的應用越來越普及.由于能夠在壓力條件下獲得樣品在三維空間中的衍射信息分布以及數據具有高信噪比等優勢,單晶XRD實驗方法不僅可以用于壓力條件下的晶體結構解析,如獲取晶胞參數、空間群、原子坐標以及原子占位等信息,而且可進一步做晶體電荷密度分析研究,得到更多的化學鍵、電荷分布及其變化等信息.本文主要介紹同步輻射高壓單晶XRD實驗方法及相關技術,其中包括單晶XRD實驗系統、單晶XRD所用DAC、單晶樣品裝填以及單晶XRD數據處理等內容.
高壓,單晶,同步輻射,X射線衍射
1 引 言
壓力作為變化范圍最大的熱力學基本參數之一[1],對物質的結構和性能有著十分重要的影響,它可以對物質幾乎所有的屬性進行調制,例如化學、結構、機械、電子、磁性以及聲子屬性等[2].在實驗室中,壓力還被用來誘導原子間發生相互作用從而產生重要的變化[3],例如從軟到硬[4,5]、從絕緣體到金屬(金屬到絕緣體)[6,7]、從鐵磁材料到超導材料[8,9]、從非晶到晶體等[10].高壓下的物質結構和性能研究已成為晶體學研究中一個十分重要和非常活躍的領域.由于晶體的物理、化學性質是由晶體的組成元素成分及其排布方式,即晶體結構決定,所以晶體結構的研究與解析工作成為晶體學研究的核心內容之一,也是物理學、化學、生命科學、地球科學等學科研究的基石.
目前,在壓力下測定晶體結構最常用的實驗手段是X射線衍射(XRD)方法.高壓XRD晶體學研究使得科學家們可以在原子水平對壓力條件下的物質開展研究,進而理解并預測物質的結構與性質[11].XRD研究使用的光源主要有X光機和同步輻射光源兩種.相對于X光機,同步輻射光源具有高能量、高亮度、低發射度、能量可調以及具有時間結構等優勢,這些優勢使得同步輻射光源在20世紀80年代之后迅速成為高壓XRD(high pressure XRD,HPXRD)實驗方法的主要光源.目前在很多同步輻射光源上都建有一條或幾條專用的HPXRD光束線(為突出主題,這里只介紹與金剛石對頂砧(DAC)技術相關的專用線站).以國際上三大高能光源為例,美國先進光源(Advanced Photon Source,APS)的Sector 13和Sector 16,歐洲同步輻射裝置(European Synchrotron Radiation Facility,ESRF)的ID-27,日本8 GeV超級光子環(Super Photon Ring-8GeV,Spring-8)的BL10 XU等,都是非常先進的以XRD方法為主的同步輻射高壓研究專用線站.
作為一種原位晶體結構研究手段,HPXRD實驗與常規XRD實驗一樣,可以采用粉末或單晶樣品.目前世界上絕大多數的HPXRD實驗站都采用粉末XRD技術.這是因為相對于單晶XRD,粉末XRD方法具有技術成熟可靠、裝置簡單、對樣品條件要求寬松、數據處理快捷、操作易于掌握等一系列優點.從目前發表的研究結果可以看出,在高壓研究中采用粉末XRD技術的實驗處于主流地位.
但與單晶XRD相比,粉末XRD實驗方法存在著衍射數據重疊、擇優取向難以避免等固有缺陷.這些缺陷不但影響了衍射峰位置信息,而且會影響衍射峰強度信息,從而對后續結構解析和精修工作產生了嚴重影響,甚至導致失敗.在高壓條件下,粉末XRD實驗方法的缺點更為明顯,例如:1)由于靜水壓條件惡化,高壓下粉末XRD的衍射峰會因為壓力梯度的增加而逐漸展寬,易導致衍射峰的重疊;2)在高壓條件下,樣品擇優取向問題會更為嚴重;3)由于在高壓下樣品變薄等原因,會導致粉末XRD衍射實驗中信噪比較差.由于這些固有不足,導致在高壓條件下的粉末XRD數據質量很難達到Rietveld精修的要求,無法完成復雜晶體結構的定量分析,對高壓環境下電荷密度[12]及化學鍵性質的變化等研究也就更無從談起.相對于粉末XRD,高壓單晶XRD不但能夠獲得樣品在三維空間的衍射信息,可以更加敏銳地捕捉到相變發生時刻的衍射峰的劈裂或融合變化,而且由于高壓單晶XRD具有數據信噪比高等優勢[13],會極大地提高衍射數據的可靠度.因此高壓單晶XRD數據不僅可以用于常規晶體結構解析和修正,而且可進一步做晶體電荷密度分析研究,得到更多的化學鍵、電荷分布及其變化等信息,為分析研究材料的物理化學性質提供幫助.
本文主要基于北京同步輻射裝置(Beijing Synchrotron Radiation Facility,BSRF)4W 2光束線高壓實驗站的單晶XRD實驗系統,對高壓單晶XRD實驗方法進行介紹,其中包括單晶XRD實驗系統、單晶XRD所用DAC、單晶樣品裝填以及單晶XRD數據處理等內容.
2 高壓單晶XRD實驗方法
2.1 單晶XRD實驗平臺
最初的同步輻射高壓單晶XRD實驗方法使用的是能量色散X射線衍射(energy dispersive X-ray diff raction,EDXD)模式[14,15].由于EDXD模式下衍射數據的強度校正難度很大,因此只能利用單晶XRD的角度和能量信息進行晶胞參數等計算.隨著同步輻射單色光技術及二維面探測器設備的快速發展,EDXD模式已經逐漸被角色散X射線衍射(angle dispersive X-ray diff raction,ADXD)模式取代.目前已有一些同步輻射高壓衍射專用線站,如美國的APS[16],日本的Spring-8[17],歐洲的ESRF[18]、Diamond[19]和PETRAIII等[20],都建立了高壓單晶XRD實驗方法.在國內,BSRF 4W 2光束線的高壓實驗站也建立了高壓單晶XRD實驗方法[21].
實驗室中常用的X射線單晶衍射儀一般會有ω,χ,φ和2θ四個轉動軸[22],而同步輻射線站上的高壓單晶XRD實驗通常只圍繞ω軸轉動,這樣在高壓粉末衍射線站上就可以通過軟硬件的升級實現單晶XRD實驗平臺的建立.由于DAC衍射窗口的限制,高壓單晶XRD實驗中一般ω的轉動范圍是±25?—±35?之間.4W 2的單晶XRD實驗過程中常用的轉動范圍為?25?—25?.
在高壓單晶XRD實驗中,非常重要的步驟就是如何將單晶樣品準確定位到ω軸上.通常在高壓實驗站,豎直方向的ω軸是一個水平放置的ω轉臺的轉軸,一般稱為旋轉中心.ω轉臺上下各有一組平移臺,轉臺下方的為X Y方向平移臺組合,轉臺上方的為X YZ方向組合,如圖1所示,其中X方向與X射線平行,Y方向為垂直于入射同步輻射光(X射線)的水平方向,Z為豎直方向.ω轉臺下面的X Y平移臺用于調整旋轉中心位置,使其與入射同步輻射光相交.ω轉臺上面的平移臺用于調整樣品位置,使其與旋轉中心和入射光的交點重合.選用精度較高的轉臺和平移臺,單晶樣品、旋轉中心以及入射同步輻射光之間的定位精度可以好于1μm.
單晶XRD實驗平臺中二維面探測器垂直于入射同步輻射光進行放置.同步輻射光的能量通過轉動雙晶單色器改變單色光能量來掃描元素吸收邊進行較定,如Zr的K邊為17.998 keV,Mo的K邊為20.000 keV.在確定能量后,樣品(旋轉中心)到探測器的距離通過標準樣品,如CeO2或LaB6等進行標定.探測器相對于入射X射線的輕微偏轉或俯仰可以通過fi t2d軟件[23]在系統標定過程中獲得.目前同步輻射高壓實驗站多配備Pilatus3、電荷耦合器件(CCD)或Mar345作為衍射信號采集探測器.根據表1的性能比較可以看到,Pilatus3探測器掃描時間非常短,但像素尺寸較大(172μm×172μm),對單晶XRD信號的空間分辨率有一定的負面影響;而且受模塊拼縫影響,Pilatus探測器死區也比較大(如Pilatus3 1M的死區為成像面積的7.2%,Pilatus3 2M的死區達到8%).高壓單晶XRD實驗因DAC的限制,本身單晶數據的完整度和分辨率就比較有限,如果再有額外的損耗,對后續的精修處理會帶來很大困難.Mar345與CCD相比在數據讀寫速度上有較大差距,但在同等的價格條件下,Mar345探測器的有效探測尺寸可以做的比較大,如Mar345的有效探測面積直徑為345 mm,而常用CCD探測器的直徑一般只有165mm且價格更貴.Mar345在一次掃描曝光過程中獲得的數據,CCD往往要重復兩到三次才能夠獲得.此外,Mar345探測器不存在電子學自噪音干擾(暗電流)問題,相比較于CCD探測器,Mar345探測器的信號噪音要更低一些,這有助于提高衍射數據精度.基于以上原因,在高壓單晶XRD實驗方法中,大多采用Mar345作為探測器,也有部分實驗采用CCD探測器在不同位置多次曝光的方法獲取數據[24].在4W 2光束線有Pilatus3 2M和Mar345兩種二維探測器,常規的粉末HPXRD實驗中多使用Pilatus3 2M探測器,單晶XRD實驗則使用Mar345探測器.為了減少衍射譜掃描時間,Mar345可以在3450× 3450(像素尺寸100μm ×100μm,讀出時間96 s)或2300× 2300(像素尺寸150μm × 150μm,讀出時間68 s)兩種模式中進行切換.
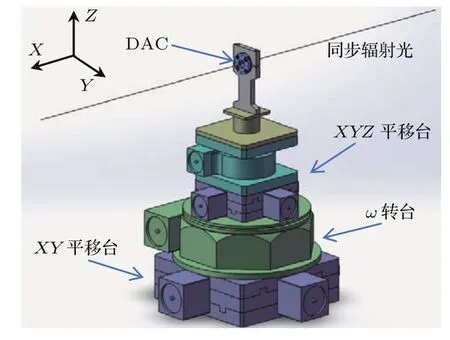
圖1 ω轉臺、平移臺、DAC與入射同步輻射光布局圖Fig.1.Layou t ofω-rotation stage,linear transition stages,DAC and synch rotron rad iation beam.
單晶XRD實驗的數據采集過程分為兩種模式:一是寬區掃描(wide scan),二是分步掃描(stepscan).寬區掃描通常會覆蓋整個實驗的ω角轉動區域,例如從?30?—30?,最后獲得一張衍射譜.寬區掃描的主要目的有兩個:首先是通過快速掃描過程確定單晶樣品的情況,如是否在實驗過程中發生樣品劈裂或結構相變等變化;其次是判斷在設定的轉動速率(轉動速率=掃描角度 /曝光時間)下進行曝光單晶衍射點強度是否合適,是否有飽和的單晶衍射點出現,然后根據曝光結果調整ω角的轉動速率.分步掃描是將整個的ω角轉動區域按某一步長進行分段掃描,例如將?30?—30?的掃描區域以2?為步長進行連續掃描和曝光,最后獲得30張衍射譜.分步掃描獲得的單晶數據會用于后續的晶體結構處理過程.分步掃描除了所需時間較多,它的主要缺點是如果有跨譜點,即衍射點出現的角度正好在相鄰兩張衍射譜的角度邊界位置,在數據后處理過程中這個衍射點的強度恢復總是比較困難.目前也可以實現根據一張寬區掃描譜完成數據的后處理,它的優點是不會有跨譜點存在,但是因為曝光時間較長,衍射譜背底噪聲比較高,衍射點的信噪比會差一些.

表1 Pilatus3 2M,Mar345以及SX-165 CCD性能比較(數據摘自MarXperts GmbH公司網站)Tab le 1.The performances comparison of Pilatus3 2M,Mar345 and SX-165 CCD(the data are taken fromthe MarXperts GmbHwebsite).
在衍射譜曝光過程中,有以下3點必須注意:1)ω轉臺電機轉動的速度必須均勻,每張衍射譜曝光時間的控制或記錄要求非常準確;2)衍射點的強度與入射X射線強度呈線性關系,因此在曝光過程中要用電離室或其他監視設備記錄入射同步輻射光的強度變化,以用于后處理過程中的衍射點強度歸一化;3)在分步掃描中,邊界角度的確定越準確越好,減少數據的空白或重疊角度.XRD系統的布局如圖2所示,沿同步輻射光傳輸方向一般為電離室、機械快門、DAC、探測器.其他設備如轉臺、K-B聚焦鏡以及限光微孔等未在圖2中表示.實驗過程中探測器處于常開狀態,通過快速的機械快門(如Vincent Associates公司的XRS6快門)控制曝光時間.為了消除ω轉臺的轉動空程,以及等待電機轉動平穩后再進入曝光區間,一般會在曝光開始角度之前設置一定的冗余行程.

圖2 高壓單晶XRD系統布局圖Fig.2.Schematic of high pressure single-crystal system.
如圖3所示,一個完整的采譜過程一般包括以下步驟:1)控制軟件命令ω轉臺轉動到起始角度θ1(起始角度=曝光開始角度?冗余行程),此時時間為t0;2)當轉臺從θ1開始加速轉動后,控制程序會快速讀取ω轉臺的角度位置(4W 2光束線單晶XRD系統常用讀取間隔<10 ms),ω轉臺在t1時刻加速完畢進入勻速轉動;3)t2時刻ω轉臺到達設定的開始曝光角度θ2時機械快門打開,X射線照射在單晶樣品上,探測器開始記錄單晶衍射信號;4)在t2時刻快門打開的同時,控制程序內部計時器開始計時,并按照固定頻率?t(如每1 s讀一次)讀取電離室強度信息進行記錄;此時程序依然保持快速讀取電機角度位置;5)t3時刻ω轉臺轉動到掃描終止位置θ3,機械快門閉合,探測器停止采譜,控制程序內部計時器停止計時,ω轉臺開始減速,控制程序將計時結果T及曝光過程中記錄的N次電離室強度平均值寫入相應的log文件保存;6)控制程序發送掃描命令,將探測器上的衍射數據保存到本地.

圖3 高壓單晶XRD數據采集過程時序圖Fig.3.Time sequence of high pressure single-crystal XRD experiment.
為了降低用戶對實驗流程控制的難度,4W 2實驗站開發了專用的控制軟件HPSXRD用于高壓單晶XRD實驗.此軟件中包括了實驗參數設定、掃描模式選擇(寬區掃描或分步掃描)、掃描時間選擇、掃描范圍及步長設定、角度讀取、快門控制、電離室讀取等功能.除了對系統硬件的控制,數據后處理部分也包括在了軟件中.利用此軟件可直接完成數據處理到hkl文件輸出的過程.圖4是HPSXRD軟件的界面及功能區說明.

圖4 HPSXRD軟件界面Fig.4.Screenshot of HPSXRD software.
2.2 高壓單晶衍射所用DAC
用一對壓砧對樣品擠壓以獲得高壓環境[25]是最常用的壓力獲得方式.金剛石因其最高硬度、最高熱導率、超高熔化溫度以及光學透明等卓越的性能優勢,成為最常用的壓砧材料.自從1959年Jamison等[26]和W ire等[27]分別提出用兩塊相對的圓錐形金剛石壓砧擠壓樣品以產生高壓強之后,DAC迅速成為高壓研究的重要設備.基于金剛石對頂砧這一基本構型,不同結構的加壓裝置也被提了出來,如NBS壓機[28],Basset壓機[29],Mao-Bell壓機[30]和Syassen-Holzapfel壓機[31]等.
除了金剛石壓砧要有足夠好的支撐以便獲得高壓環境外,高壓單晶XRD所用DAC與粉末XRD所用DAC的最大區別,就是還需要有盡量大的衍射張角,以覆蓋足夠大的倒易空間范圍.1974年,Merrill-Basset DAC[32]的出現對高壓單晶XRD實驗方法的推廣起到了重要的作用.Merrill-Basset DAC十分小巧而且結構簡單,衍射張角也足夠大,非常適于安裝在實驗室衍射儀的測角頭上.除了Merrill-Basset DAC,也有其他一些DAC[33?40]可以用于高壓單晶研究.為了獲得足夠大的衍射窗口,使衍射數據覆蓋盡量大的倒易空間,這些DAC的設計過程一般都采用兩種方式.第一種是沿加壓軸方向的軸向衍射方式.這種方式多使用鈹(beryllium)作為墊塊材料[33,35,36,40],單晶衍射信號穿過鈹墊塊后到達探測器.鈹墊塊的優勢是對單晶衍射數據的吸收非常弱,能夠使衍射窗口不受墊塊開孔的限制;但鈹的存在依然會增加衍射背底,同時會產生附加的衍射環[41];鈹材料氧化后的毒性問題也會帶來使用和加工成本的提高.第二種是垂直于加壓軸的側面衍射方式[37?39,42],這種方式可以獲得足夠大的衍射窗口,覆蓋更大的倒易空間,但是單晶衍射信號穿透路徑比較復雜,有可能會穿過封墊、壓砧、樣品等多種材料,進行強度修正時難度較大.
Bohler在2004年提出來一種新的Bohler-Almax壓砧結構[43,44],在這種新結構中,金剛石壓砧大臺面端的側腰被加工成圓錐形,并用具有相應下凹圓錐形開孔的碳化鎢墊塊進行支撐.這種壓砧-墊塊組合用下部的斜側面支撐代替了傳統的底部支撐,能有效減少硬質合金墊塊對衍射窗口的限制,在高壓單晶XRD實驗中具有很大的優勢;同時,它還能保證支撐面與壓砧砧面之間的面積比足夠大,使得金剛石壓砧能夠達到足夠高的壓力范圍.Ahsbahs[45]在2004年也提出過類似的結構.將Bohler結構壓砧裝入Merrill-Basset DAC[41]或者BX 90 DAC[46]中,可以獲得足夠大的衍射窗口(最大約90?),對提高單晶XRD數據的完整度與精度有著非常大的幫助.
2.3 樣品裝填
在高壓實驗中,樣品準備是一件非常復雜的工作,高壓單晶XRD實驗尤其如此.在單晶樣品裝填的前期準備工作中不但要考慮DAC的適用性,金剛石壓砧砧面尺寸與封墊材料是否符合實驗壓力范圍要求,還要考慮封墊預壓厚度、樣品孔直徑、樣品尺寸(包括厚度)以及傳壓介質選擇等一系列條件.即使各種因素都考慮到了,如何把單晶樣品按照預設的方式完美的置于樣品腔中,也是一項非常有挑戰性的工作.
這里我們不介紹通過加溫[47]或加壓[36,48]方式在樣品腔中合成/生長出單晶顆粒的樣品準備方式,只涉及最常用的單晶顆粒裝填方式.圖5是比較典型的單晶樣品裝填方式,根據里面相應的組分,下面分項進行簡單介紹.

圖5 典型的DAC樣品腔單晶樣品裝填布局Fig.5.Atypical single-crystal sample con figu ration in sample chamber of DAC.
2.3.1 封 墊
一般來說,在確定了實驗可能會達到的壓力范圍之后,我們會選擇相應砧面直徑的DAC以及封墊材料.常用的封墊材料主要有硬質合金T301、錸和鎢等,壓力范圍不高的實驗多選用T301.對于從側面接受衍射信號的單晶實驗,也會選用鈹[49,50]和非晶硼[51,52]作為封墊材料.封墊的作用除了提供樣品腔以封閉樣品和傳壓介質等提供(準)靜水壓環境,還可以在高壓下對金剛石砧面周圍形成機械支撐,防止砧面尖端由于巨大的剪切應力而損壞.在封墊使用之前需要用壓砧進行預壓以增加硬度,然后再在預壓位置的中心鉆出樣品孔.預壓后的封墊厚度一般在30—40μm,通常而言,實驗壓力范圍越高,封墊預壓厚度越小.如果選擇填充液體傳壓介質,樣品孔的直徑一般不超過壓砧砧面直徑的40%.
2.3.2 傳壓介質
在高壓單晶XRD實驗中,樣品所處的靜水壓或準靜水壓環境的好壞對衍射數據的質量非常重要.在較高壓力條件下樣品腔內軸向與徑向壓力差會隨著壓力上升而逐漸增加,這往往會導致單晶衍射點的形狀發生畸變,對后期強度校正工作帶來不利影響.由于不存在粉末XRD實驗中樣品顆粒之間的作用力,單晶XRD實驗中非靜水壓問題帶來的影響要稍微好一些.一般來說,小的X射線入射光斑和小粒度的單晶樣品顆粒會減弱非靜水壓的不利影響,但傳壓介質的選擇依然是非常重要的影響因素.
在高壓實驗中,當單晶樣品被流體狀態的傳壓介質包圍時,它處于一個各向均勻受力的狀態,這時為靜水壓條件.隨著壓力的上升,流體狀態的傳壓介質會逐漸固化,靜水壓條件被破壞,樣品在各個方向所受的壓力也不再均勻,樣品腔內壓力不均勻性以及壓力梯度都會逐漸增加.在實驗中選用的傳壓介質和實驗壓力范圍有直接關系.在最常用的幾種液體傳壓介質中,4:1的甲醇、乙醇混合溶液[53],或者16:3:1的甲醇、乙醇和水的混合溶液在10 GPa時依然能提供較好的靜水壓環境,但隨后靜水壓條件就會迅速下降[54].硅油與甲醇、乙醇混合溶液類似,它能在12 GPa左右也提供較好的靜水壓環境[55,56],但硅油在衍射信號中產生的背底噪音會略高一些.Ar可以通過低溫液化方式充入樣品腔內,是目前最常用的氣體傳壓介質.300 K條件下Ar在1.5 GPa固化,它能夠達到的準靜水壓范圍為11GPa左右[14].室溫條件下Ne在2.4GPa固化,它的準靜水壓上限能達到20 GPa[57].He的固化壓力在11 GPa以上[58],毫無疑問它是最好的靜水壓傳壓介質,甚至在100 GPa的壓力環境下也可以提供較好的準靜水壓條件[59].Ne和He一般只能用高壓充氣[60,61]的方法注入樣品腔中.
在選擇傳壓介質時,除了要考慮實驗的靜水壓范圍,還要考慮傳壓介質是否會與樣品發生反應.另外在He,Ne等小分子作為傳壓介質時,還要考慮它們是否會在壓力條件下滲入樣品的結構中,特別是對一些晶胞結構比較大的樣品.
2.3.3 單晶樣品尺寸
單晶顆粒形狀與尺寸的選擇在高壓單晶XRD實驗的樣品準備過程中同樣是一個很重要的問題.最理想的單晶樣品是球形,這樣在實驗過程中將樣品放置在X射線與旋轉中心的交點位置后,樣品參與衍射的體積變化、樣品對入射X光及單晶衍射信號的吸收可以認為保持不變,后處理過程中相應的校正可以略去不做.通常來說,在樣品準備過程中會盡量選擇或切割出形狀比較規則的單晶顆粒,如正方體、長方體或圓柱體等,這主要是為了降低在后處理過程中如果需要進行上述校正時的難度.在樣品準備過程中,除了樣品的長度與寬度不能超過樣品孔直徑,還有以下兩點需要特別注意.
首先,單晶樣品的厚度不能超過封墊的預壓厚度,這樣才能讓樣品完全被傳壓介質包圍,使其處于靜水壓環境中.同樣,在封墊預壓厚度與樣品厚度之間需要保留一部分的余量,以免在加壓過程中因為樣品孔變薄使樣品直接被兩個壓砧砧面擠壓,從而導致靜水壓環境被破壞,甚至導致單晶顆粒破碎.
其次,單晶顆粒在垂直于DAC加壓軸方向的長度與寬度尺寸,要與入射X光的尺寸結合考慮.對于不同尺寸的入射X光,單晶尺寸的選擇會有所不同.以BSRF 4W 2高壓實驗站為例,在進行高壓單晶XRD實驗時,樣品處X射線半高全寬為50μm×50μm,一般單晶樣品的水平方向尺寸在20—30μm,垂直方向可大可小.這樣在實驗過程中當單晶樣品進行轉動時,樣品參與衍射的體積保持不變,在后續數據處理過程中可以略去樣品體積校正的工作.如果實驗站的X射線光斑尺寸比較小,如APS的HPCAT16BM-D,它在樣品處的X射線光斑半高全寬為5μm×5μm.這種情況下如果要求樣品在實驗過程中始終被X射線覆蓋難度比較大,一般盡量選擇形狀比較規則的樣品,以降低后續數據處理中的難度.但小光斑也有其他優勢,特別是對多顆粒樣品的單晶衍射有著非常大的幫助[47,62].
需要特別說明的是,對于密度較大的單晶樣品,在進行衍射信號強度處理過程中必須要考慮樣品對X射線吸收帶來的影響.
2.4 數據處理
相對于高壓粉末XRD實驗,單晶XRD實驗的數據處理過程更為繁瑣,涉及的內容也較多.為了降低用戶在單晶數據后處理工作中的難度,我們在開發HPSXRD軟件的過程中集成了單晶數據處理的主要功能模塊.圖6是數據處理的一般流程,下文按照這一流程進行介紹.hkl文件輸出之后,可以用于樣品結構精修或電荷密度分布(electron density distribution,EDD)計算等研究內容.
2.4.1 衍射數據的指標化
有時因為準備過程中的問題,用戶的樣品包含有幾個單晶顆粒,或者由于在加壓過程中樣品的變化,導致單晶樣品碎裂.在這種情況下,所采集的衍射數據不再是單晶衍射,而是有限顆粒(多顆粒)衍射,即有多套衍射譜疊加在一起的衍射數據.直接用以往的方法,如Niggli約化法[63,64]或傅里葉變換法[65,66],處理這種數據便遇到了困難.為此,我們發展了一套新的數據處理流程:首先用粉末XRD指標化方法確定晶體的晶胞參數;然后再用遺傳算法嘗試指標化相應的衍射點數據.這種算法會不斷調整模型方位角來匹配所得到衍射數據,直到找到一個最佳匹配解.通過此解就可以計算出定位矩陣(UB矩陣).最后,在得到最優化解以后,程序首先將衍射點最多的小塊單晶的數據提取出來,然后反復利用遺傳算法和已經確定的晶胞參數繼續對剩余的衍射數據進行指標化,這樣可以完成對所有有效衍射數據的指標化過程[67].
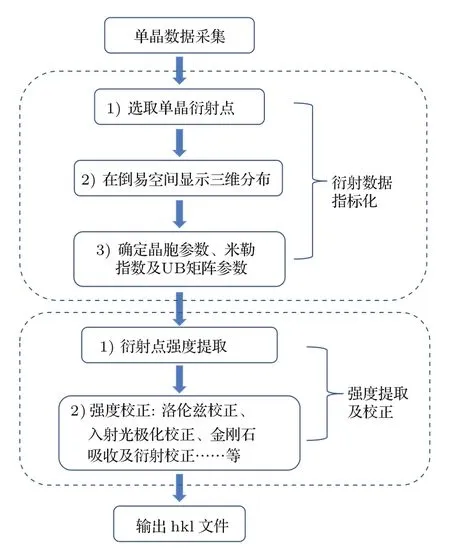
圖6 高壓單晶XRD數據處理的一般流程Fig.6.High pressu re single-crystal XRD data processing procedure.
上述功能已經集成到HPSXRD軟件中的Index模塊當中,該模塊既可以完成單個單晶顆粒樣品的指標化工作,也可以對多個單晶顆粒樣品分別進行指標化.
2.4.2 衍射點強度的提取
單晶衍射點強度的提取是單晶XRD實驗中重要的環節.隨著目前二維面探測器的普及,如何準確、可靠地從二維衍射數據中提取衍射點凈強度更是一個重要問題.經過近二十多年的發展,目前已經建立了一些比較可靠的方法.這些方法可以分為兩類.一類是峰形函數法,使用二維Gaussian或Lorentz函數做峰形函數來擬合衍射點形狀,然后對擬合得到的方程作積分得到相應衍射點的強度[68].這種方法的優點是不管衍射信號強弱得到的衍射強度都比較精確,且不會出現負衍射強度的情況;缺點是如果衍射點形狀不規則,則提取的衍射強度偏差較大.而另一類處理衍射點強度的方法是衍射峰-背底區域劃分法,它是利用統計學方法對一定像素面積內的衍射點強度進行分析,并確定衍射峰值、背底值及凈衍射強度[69].這一方法的優點是可以處理因為應力導致的不規則形狀衍射斑點,缺點是對于比較弱的衍射點,凈衍射強度值有可能出現負值,且如果兩個衍射峰距離較近(峰重疊)時,該方法失效.理論上單晶衍射不會發生衍射峰重疊情況,但是在高壓單晶衍射過程中,由于晶體破碎或受金剛石單晶壓砧衍射影響有可能會出現這種情況.
考慮到在壓力較高時,晶胞內部應力較大會導致衍射斑形狀不再是Gaussian分布(見圖7不同壓力下同一衍射點的形狀對比),在HPSXRD軟件里使用衍射峰-背底劃分法作為衍射強度提取方法.具體算法參考了Bruker公司早期的CAD4型四圓單晶衍射儀點探測器一維衍射數據提取算法,過程如下.

圖7 高壓單晶XRD實驗中由于非靜水壓導致的衍射點形變(圖中為同一衍射點在不同壓力下的形狀;樣品為Cr2O3單晶,傳壓介質Ne) (a)采譜壓力為6.6 GPa;(b)采譜壓力為50.7 GPaFig.7.The deformation of diff raction point caused by non-hyd rostatic conditions(single-crystal sample is Cr2O3,pressu re med iumis Neon):(a)6.6 GPa,hyd rostatic condition;(b)50.7 GPa,non-hyd rostatic condition.
首先,劃定包含衍射點在內N個像素的一小塊范圍.然后,計算該范圍內的平均光子計數強度A1:

其中Pi表示第i個像素點的光子計數,N表示所選范圍內的像素數目.
其次,計算所選范圍內,光子數小于A1的所有像素的平均計數值A2:
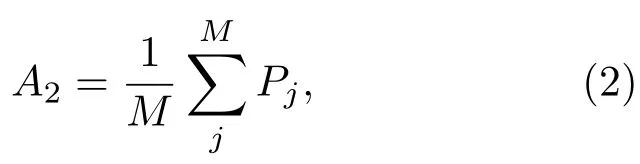
其中Pj表示第j個像素的光子計數,M表示所有光子數小于A1的像素的數目.
最后得到衍射點的強度為

將上述方法擴展,就可以處理二維衍射數據.首先確定含有衍射峰的一個nx×ny像素(nx,ny可以調整,通常設為nx=ny)區域.然后按上述公式就可以將該區域內衍射峰和背底的區域劃分出來.
完成衍射點強度提取后,只是得到了直接探測到的衍射信息,還需要對其進行洛倫茲校正、光源極化校正、金剛石吸收與衰減校正、掃描時間校正以及入射光強校正,最終得到可以直接用于結構精修的準確的強度信息.在HPSXRD程序中的Intensity模塊可以用于衍射點強度提取與校正工作.下文針對前面三項校正流程進行介紹.掃描時間及入射光強校正主要是針對步進掃描過程的曝光時間及入射同步輻射光強歸一化,因為涉及內容比較簡單,不做過多說明.
2.4.3 洛倫茲校正
在數據采集過程中,晶體繞樣品臺豎直軸ω軸以均勻角速度轉動,因為倒易空間中不同倒易點距轉軸的距離r不同,它們在經過衍射球(埃瓦爾德球,Ewald sphere)的線速度v也不同[70].因為線速度的差別,實驗過程中不同等效點系的衍射點具有不同的曝光時間,洛倫茲校正就是對這種直接測量的衍射點做線性校正,校正后的衍射點強度為:Icorr=Iobs×(Y/D′),其中Iobs表示直接測量得到的衍射點強度,Y表示探測器上的衍射點M的Y軸坐標值,D′表示樣品到探測器上衍射斑M的距離,D′=(D2+Y2+Z2)1/2,如圖8所示.圖8中O點是位于埃瓦爾德球球心的樣品原點(衍射儀坐標系);O′為倒易點陣原點,坐標(1/λ,0,0),1/λ為埃瓦爾德球半徑,等于O 和O′之間的距離;O′′為直通X射線在探測器上的位置,它的坐標為(D,0,0),其中D為樣品到探測器的距離;r為某一與埃瓦爾德球相交的倒易點m到ω轉軸的半徑;v為衍射點m的線速度,v=r×ω;M為m在探測器上的衍射點,它的坐標為(D,Y,Z).
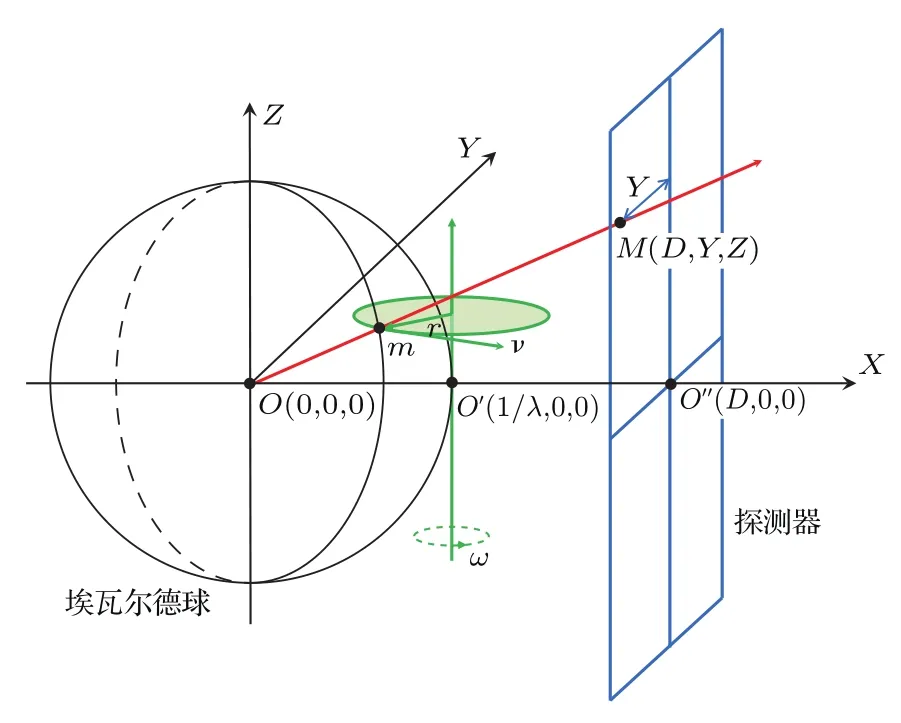
圖8 洛倫茲校正原理圖Fig.8.Schematic of Lorentz correction.
2.4.4 極化校正
一般實驗室X射線衍射儀的光源為非極化光,因此對于衍射強度的校正只考慮單色器引起的極化效應即可.但是同步輻射光源為極化光,因此,同步輻射的衍射數據在考慮極化效應時,既要考慮單色器極化因素,也要考慮光源極化因素.對于入射光的極化校正,Kahn等[71]已經發展了相應的方法,HPSXRD程序中也使用了一樣的校正方法,下面對計算方法進行簡單介紹.

布拉格衍射點強度校正為:Icorr=Iobs/P,式中,Icorr表示校正后的衍射強度;Iobs表示原始衍射強度;P代表極化因子,由兩部分組成,其中,P0為單色器極化,P′是光源極化,θ表示布拉格衍射角,?表示衍射點在探測器上的坐標位置與探測器水平坐標的夾角(如圖9所示),ζ表示同步輻射光源的極化參數,其變化范圍為?1—1之間,可以從光束線參數或者對探測器所測背景擬合得到.經過測試,4W 2的極化參數為?0.7.

圖9 光源極化校正中角θ和?角位置示意圖,其中M代表單晶衍射點Fig.9.Diff raction geometry and parameters of polarization correction.
2.4.5 金剛石壓砧的吸收和衍射校正
在高壓單晶XRD實驗過程中,不可避免地要使用到大塊金剛石單晶作為壓砧.大塊的金剛石單晶一方面會對入射同步輻射光產生衍射,另一方面還有對入射光的吸收衰減作用.圖10為DAC中上游(入射側)金剛石壓砧在不同旋轉角度時透過的同步輻射光強度變化.從圖10可以看到,在一些角度透射光會有劇烈的強度變化,這種強度變化是由金剛石衍射導致的,它會嚴重影響樣品衍射信號的強度.而金剛石吸收帶來的入射光強度變化可以從透射光強度包絡線上看出來,它導致的強度變化相對要平緩一些,而且只和轉動角度相關.在單晶實驗之前或結束之后,需要對上游金剛石在不同角度對入射同步輻射光的強度影響進行記錄.在后處理的衍射強度校正過程中,HPSXRD程序會根據導入的金剛石校正文件自動對單晶衍射點進行強度校正,以保證最終能夠得到正確的結構因子的強度值.
DAC內的下游(衍射側)金剛石對單晶樣品的衍射信號強度也有影響.但因為測量與分析的難度,目前只通過程序中的路徑擬合對吸收衰減校正,沒有考慮衍射造成的強度偏差.

圖10 上游金剛石透過光強隨角度的變化Fig.10.Intensity of the transmitted X-ray going through the upstreamd iamond anvil at variousωangles.
3 應用舉例
在高壓單晶XRD實驗平臺測試過程中,我們用ZnO單晶進行了測試,并根據單晶數據獲得了壓力條件下ZnO的EDD結果.實驗中DAC的砧面直徑為250μm,封墊材料為T301,封墊預壓厚度30μm,樣品腔直徑100μm.單晶樣品大小為(寬)22μm × (高)40μm × (厚)23μm.壓力標定采用紅寶石熒光法[72],傳壓介質為Ne.實驗過程中ω角轉動范圍為?27?—27?,采用步進掃描模式,每2?收集一張衍射譜,每張衍射譜曝光時間為30 s.使用HPSXRD程序對數據進行處理,在輸出hkl文件之后,用Shelx-97程序[73]進行結構精修,表2為1.5 GPa和4.7 GPa時ZnO單晶衍射數據得到的結構精修結果.圖11是這兩個壓力條件下ZnO的EDD情況及差值EDD圖,EDD結果由最大熵算法[74]得到.從圖11(c)可以看到:隨著壓力的增加,電荷分布從Zn和O的核部向殼層遷移,使得Zn—O成鍵區電荷密度增加;而且由于Zn—O為不等性sp3雜化,使得成鍵區電荷增加不一致.
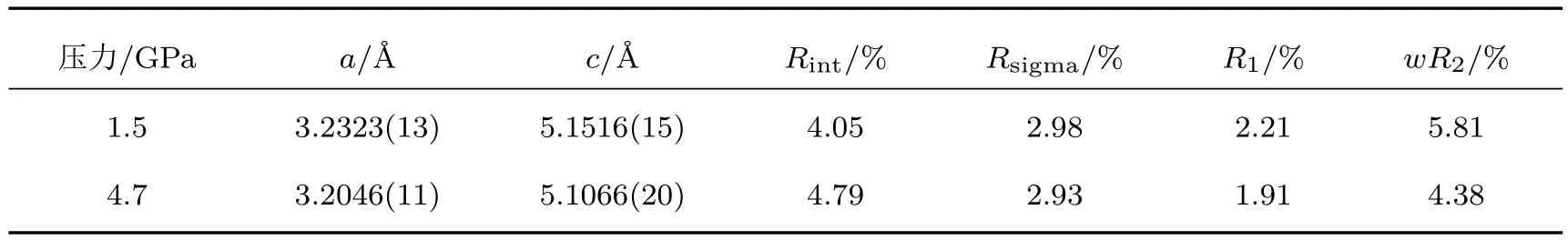
表2 ZnO在不同壓力下的精修結果Tab le 2.The refinement results of ZnOunder diff erent pressure.
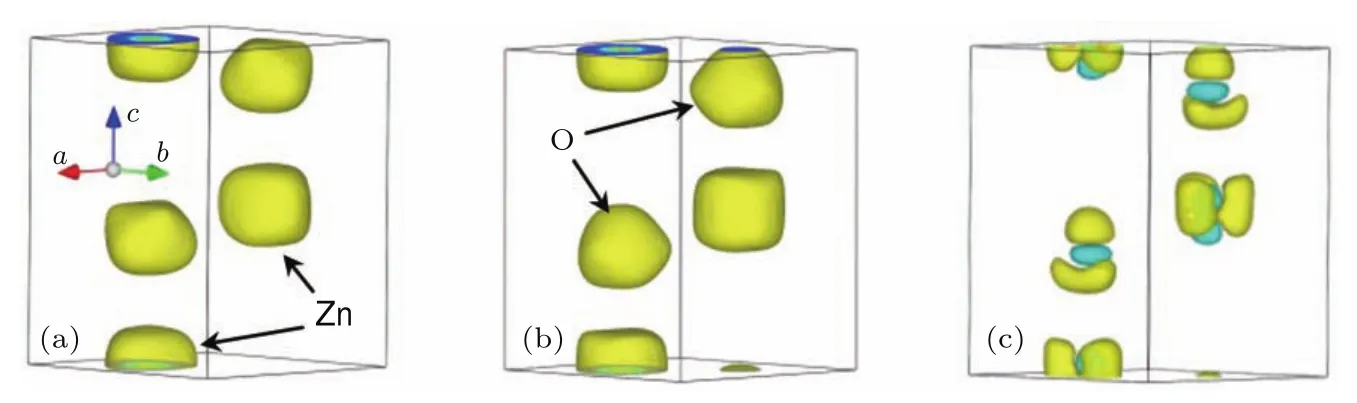
圖11 ZnO在不同壓力下的電荷密度分布結果及差值電荷密度 (a)1.5 GPa,電荷密度分布;(b)4.7 GPa,電荷密度分布;(c)差值電荷密度,圖中黃色表示為電荷密度增加區域,青色表示電荷密度減少區域Fig.11.The EDD restored of single-crystal ZnOusing maximumentropy method:(a)EDD resu lts under 1.5 GPa;(b)EDD results under 4.7 GPa;(c)diff erence EDD results.
4 總結與展望
本文介紹了同步輻射高壓單晶XRD實驗方法,其中重點針對同步輻射角色散高壓單晶XRD實驗系統的設定、單晶XRD所用DAC的發展、單晶樣品裝填的注意事項、數據后處理的主要步驟等內容進行了說明,并在最后以ZnO為例對高壓單晶XRD的應用進行了介紹.
得益于高壓實驗技術以及同步輻射光源的不斷發展,高壓單晶XRD實驗方法已經成為很多高壓線站配備的實驗手段.隨著探測器技術的不斷發展,成本適中、面積較大、無死區或死區很小、掃描時間非常短的探測器必將會在線站中逐漸普及,這對提高單晶XRD實驗的效率將起到極大的推動作用.
目前世界上的主要高能光源,如APS,ESRF和Spring-8都在進行升級計劃,中國也計劃在十三五期間建設能量為6 GeV的新的高能光源.這些光源無一例外都把獲得更小發射度的同步輻射光作為追求目標之一.隨著發射度的降低,微米及亞微米聚焦光斑將成為高壓線站常規的實驗條件,這將推動高壓多顆粒XRD實驗技術的發展與普及.多顆粒XRD實驗技術基于單晶XRD發展而來,它的樣品可能是幾十或者幾百顆微單晶組成.多顆粒的樣品形態可以很好地彌補高壓單晶XRD實驗中數據冗余度與完整度不足的缺陷,這種方法在晶體學研究中具有非常大的潛力和應用前景.
感謝中國科學院高能物理研究所劉景研究員、李延春博士和白利剛博士在本文寫作過程中的建議,感謝楊棟亮博士幫助完成圖1的繪制工作;特別感謝美國華盛頓卡內基研究院沈國寅教授在4W 2高壓單晶XRD系統搭建過程中的建議與幫助.
[1]Hemley R J 2000Annu.Rev.Phys.Chem.51 763
[2]Shen G,MaoHK2017Rep.Prog.Phys.80 16101
[3]Du ff y TS 2005Rep.Prog.Phys.68 1811
[4]Huang Q,Yu D,Xu B,Hu W,Ma Y,W ang Y,ZhaoZ,W en B,He J,Liu Z,Tian Y 2014Nature510 250
[5]W ang L,Liu B,Li H,Yang W,Ding Y,Sinogeikin S V,Meng Y,Liu Z,Zeng X C,MaoW L 2012Science337 825
[6]Duan D,Liu Y,Tian F,Li D,Huang X,ZhaoZ,Yu H,Liu B,Tian W,Cui T2014Sci.Rep.4 6968
[7]Ma Y,Eremets M,Oganov AR,X ie Y,Trojan I,Medvedev S,Lyakhov AO,Valle M,Prakapenka V 2009Nature458 182
[8]Sun L,Chen X J,GuoJ,GaoP,Huang Q Z,W ang H,Fang M,Chen X,Chen G,W u Q,Zhang C,Gu D,Dong X,W ang L,Yang K,Li A,Dai X,MaoH,ZhaoZ 2012Nature483 67
[9]Kang D,Zhou Y,Y iW,Yang C,GuoJ,Shi Y,Zhang S,W ang Z,Zhang C,Jiang S,Li A,Yang K,W u Q,Zhang G,Sun L,ZhaoZ 2015Nat.Commun.6 7804
[10]Zeng Q,Sheng H,D ing Y,W ang L,Yang W,Jiang J Z,MaoW L,MaoHK2011Science332 1404
[11]Katrusiak A2008Acta Crystallogr.Sect.A64 135
[12]Katrusiak A2004Acta Crystallogr.Sect.A60 409
[13]Ballaran TB,Ku rnosov A,Trots D 2013High Press.Res.33 453
[14]Loubeyre P,LeTou llec R,Hausermann D,Han fl and M,Hemley R J,MaoHK,Finger L W 1996Nature383 702
[15]MaoHK,Jephcoat AP,Hemley R J,Finger L W,Zha C S,Hazen R M,Cox D E 1988Science239 1131
[16]Shen G,Sinogeikin S 2015Rev.Sci.Instrum.86 71901
[17]Ohishi Y,HiraoN,Sata N,Hirose K,Takata M2008High Press.Res.28 163
[18]Mezouar M,C richton W A,Bauchau S,Thu rel F,W itsch H,Torrecillas F,Blattmann G,Marion P,Dabin Y,Chavanne J,Hignette O,Morawe C,Borel C 2005J.Synchrotron Radiat.12 659
[19]Nowell H,Barnett SA,Christensen KE,Teat S J,Allan D R 2012J.Synchrotron Radiat.19 435
[20]Rothkirch A,Gatta G D,Meyer M,Merkel S,Merlini M,Liermann HP 2013J.Synchrotron Radiat.20 711
[21]Liu J 2016Chin.Phys.B25 076106
[22]Katrusiak A2004High-Pressure Crystallography(Dord recht:Springer Netherlands)pp57–68
[23]Hammersley AP,Svensson S O,Han fl and M,Fitch AN,Hausermann D 1996High Press.Res.14 235
[24]Dera P,Lavina B,Meng Y,Prakapenka V B2011J.Solid State Chem.184 3040
[25]Bridgman P W 1937Proc.Am.Acad.Arts Sci.71 387
[26]Jamieson J C,Lawson AW,Nachtrieb N D 1959Rev.Sci.Instrum.30 1016
[27]W eir C E,Lippincott E R,van Valkenburg A,Bunting E N 1959J.Res.Natl.Bur.Stand.A63 55
[28]PiermariniG J,Block S 1975Rev.Sci.Instrum.46 973
[29]Bassett W A,Takahashi T,Stook P W 1967Rev.Sci.Instrum.38 37
[30]MaoHK,Bell P M1978Carnegie Inst.Yearb.77 904
[31]Huber G,Syassen K,HolzapfelW B1977Phys.Rev.B15 5123
[32]Merrill L,Bassett W A1974Rev.Sci.Instrum.45 290
[33]Keller R,HolzapfelW B1977Rev.Sci.Instrum.48 517
[34]Allan D R,Miletich R,Angel R J 1996Rev.Sci.Instrum.67 840
[35]MaoHK,Bell P M1980Carnegie Inst.Yearb.79 409
[36]W eir C E,Block S,Piermarini G J 1965J.Res.Natl.Bur.Stand.C69 275
[37]Schiferl D 1977Rev.Sci.Instrum.48 24
[38]Ahsbahs H1984Rev.Sci.Instrum.55 99
[39]Malinowski M1987J.Appl.Crystallogr.20 379
[40]Chervin J C,Canny B,Besson JM,Pruzan P 1995Rev.Sci.Instrum.66 2595
[41]Moggach S A,Allan D R,Parsons S,W arren J E 2008J.Appl.Crysta llogr.41 249
[42]Bassett W A,Spetzler H,Angel R J,Chen G R,Shen AH,Reichmann HJ,Yoneda A1998Rev.High Press.Sci.Technol.7 142
[43]Boeh ler R,de Hantsetters K2004High Press.Res.24 391
[44]Boeh ler R 2006Rev.Sci.Instrum.77 115103
[45]Ahsbahs H2004Z.Für Krist.-Cryst.Mater.219 305
[46]Kantor I,Prakapenka V,Kantor A,Dera P,Ku rnosov A,Sinogeikin S,Dubrovinskaia N,Dub rovinsky L 2012Rev.Sci.Instrum.83 125102
[47]LiR 2016Ph.D.D issertation(Beijing:Institu te of High Energy Physics,Chinese Academy of Sciences)(in Chinese)[李蕊 2016博士學位論文(北京:中國科學院高能物理研究所)]
[48]Miletich R,Allan D R,Kuhs W F 2000Rev.Mineral.Geochem.41 445
[49]Macavei J,Schu lz H1990Rev.Sci.Instrum.61 2236
[50]MaoHK,Xu J,Struzhkin V V,Shu J,Hemley R J,Sturhahn W,Hu MY,AlpE E,Vocad loL,AlfèD,Price G D,G illan MJ,Schwoerer-B?hning M,H?usermann D,Eng P,Shen G,Giefers H,Lübbers R,W ortmann G 2001Science292 914
[51]MaoHK,Shu J,Fei Y,Hu J,Hemley R J 1996Phys.Earth P lanet.In ter.96 135
[52]Lin J F,Shu J,MaoHK,Hemley R J,Shen G 2003Rev.Sci.Instrum.74 4732
[53]PiermariniG J,Block S,Barnett JD 1973J.Appl.Phys.44 5377
[54]Angel R J,Bu jak M,ZhaoJ,Gatta G D,Jacobsen S D 2007J.Appl.Crystallogr.40 26
[55]Shen Y,Kumar R S,Pravica M,N icol MF 2004Rev.Sci.Instrum.75 4450
[56]Ragan D D,C larke D R,Schiferl D 1996Rev.Sci.Instrum.67 494
[57]Takemu ra K,Singh A2006Phys.Rev.B73 224119
[58]Besson J,Pinceaux J 1979Science206 1073
[59]Dewaele A,Loubeyre P,Mezouar M2004Phys.Rev.B70 94112
[60]Kenichi T,Sahu P C,Yoshiyasu K,YasuoT2001Rev.Sci.Instrum.72 3873
[61]Rivers M,Prakapenka V B,KuboA,Pu llins C,Holl C M,Jacobsen SD 2008High Press.Res.28 273
[62]Zhang L,Meng Y,Dera P,Yang W,MaoW L,MaoHK2013Proc.Natl.Acad.Sci.110 6292
[63]K?ivy I,G ruber B1976Acta Crystallogr.A32 297
[64]And rews L C,Bernstein HJ 1988Acta Crystallogr.Sect.A44 1009
[65]Steller I,Bolotovsky R,Rossmann MG 1997J.Appl.Crystallogr.30 1036
[66]Rossmann MG 2001International Tables for Crystallography Volume F:Crystallography ofBiologicalMacromolecu les(Dord recht:Springer Netherlands)pp209–211
[67]Li H,Li X,He M,Li Y,Liu J,Shen G,Zhang Z 2013J.Appl.Crystallogr.46 387
[68]Dera P,Zhuravlev K,Prakapenka V,RiversML,Finkelstein G J,G rubor-Urosevic O,Tschauner O,C lark SM,Downs R T2013High Press.Res.33 466
[69]Leslie AG W 2001In ternational Tables for Crystallography Volume F:Crystallography of Biologica l Macromolecules(Dord recht:Springer Netherlands)pp212–217
[70]Holton J M,Frankel KA2010Acta Crystallogr.Sect.D66 393
[71]Kahn R,Fou rme R,Gadet A,Janin J,Dumas C,And re D 1982J.Appl.Crysta llogr.15 330
[72]MaoHK,Xu J,Bell P M1986J.Geophys.Res.Solid Earth91 4673
[73]Sheld rick G M2008Acta Crystallogr.Sect.A64 112
[74]Collins D M1982Nature298 49
PACS:62.50.–p,07.85.Qe,61.05.cpDOI:10.7498/aps.66.036203
High pressu re single-crystal synch rotron X-ray d iff raction techn ique?
Li Xiao-Dong1)?Li Hui2)Li Peng-Shan1)
1)(Center for Mu lti-d isciplinary Research,Institu te of High Energy Physics,Chinese Academy of Sciences,Beijing 100049,China)2)(Institu te ofMicrostructure and Properties of Advanced Materials,Beijing University of Technology,Beijing 100124,China)(Received 10 January 2017;revised manuscript received 13 January 2017)
Alot ofgreatwork hasbeen done since thehigh pressure research carried out on synchrotron radiation facility almost 40 yearsago.The history ofhigh pressure single-crystaldiff raction research on synchrotron radiation facility hasalsobeen more than 20 years.Recently,with the development of synchrotron X-ray optical techniquesand high pressure technology,especially the invention and improvement of large opening diamond anvil cell(DAC),high pressure single-crystal X-ray diff raction(HPSXRD)method has become more and more popular in high pressure studies.The HPSXRD can be used toperformstructure determination and refinement toobtain the information about lattice parameter,space group,atomic coordinate and site occupation.Compared with powder X-ray diff raction,the HPSXRD can not on ly obtain the three-dimensional diff raction in formation of samples,but alsohavemuch better signal-to-noise ratio.Furthermore,the HPSXRD data can be used tostudy the electron density distribution toobtain more information about chemical bonds and electron distribution.In this work,we introduce the HPSXRD method in synchrotron radiation facilities,including the knowledge of single-crystal X-ray diff raction experimental system,DAC for HPSXRD,sample loading,and HPSXRD data processing.
high pressure,single-crystal,synchrotron radiation,X-ray diff raction
10.7498/aps.66.036203
?國家自然科學基金(批準號:11274030,11474281)資助的課題.
?通信作者.E-mail:lixd@ihep.ac.cn
*Project supported by the National Natural Science Foundation of China(G rant Nos.11274030,11474281).
?Corresponding author.E-mail:lixd@ihep.ac.cn
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中國纖檢(2022年8期)2022-09-22 07:28:06
紡織標準與質量(2022年2期)2022-07-12 06:12:50
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
質量技術監督研究(2019年1期)2019-04-25 12:27:40
中國纖檢(2017年8期)2017-12-20 21:18:34
中國纖檢(2017年7期)2017-12-15 13:04:41
爆炸與沖擊(2017年3期)2017-06-07 08:21:19
發明與創新(2016年38期)2016-08-22 03:02:52
