高頻熔融-X射線熒光光譜法測定擬薄水鋁石中多種微量和痕量雜質成分
2017-09-03 10:31:37馬兵兵
濕法冶金 2017年4期
馬兵兵
(重慶市計量質量檢測研究院,重慶 400020)
高頻熔融-X射線熒光光譜法測定擬薄水鋁石中多種微量和痕量雜質成分
馬兵兵
(重慶市計量質量檢測研究院,重慶 400020)
研究了采用高頻熔融-X射線熒光光譜法測定擬薄水鋁石中SiO2、Fe2O3、Na2O、K2O、CaO、ZnO、TiO2、V2O5、P2O5等微量和痕量雜質含量。樣品以四硼酸鋰-偏硼酸鋰混合熔劑熔融,以溴化鋰為脫模劑,分別在800 ℃和1 000 ℃下加熱3 min,之后在1 150℃下熔融8 min,冷卻后制成玻璃片,進行測定。用與擬薄水鋁石基體及雜質含量相近的氧化鋁和氫氧化鋁國家標準物質繪制校準曲線,在相同條件下熔融成玻璃片測定各成分熒光強度。當雜質質量分數大于0.001%時,本法測定值與ICP-AES法和AAS法測定值相近,9種雜質成分檢出限在0.000 084%~0.005 6%之間,不同成分測定值的相對標準偏差(n=10)在0.36%~8.3%之間。
高頻熔樣;X射線熒光光譜法;擬薄水鋁石;微量;痕量;雜質
擬薄水鋁石(α′-AlOOH·nH2O,n=0.08~0.62)又稱一水合氧化鋁或假一水鋁石,無毒、無味、無臭,濕品為白色膠體狀,干品為粉末狀或結晶體。擬薄水鋁石廣泛用于催化劑載體、活性氧化鋁原料,也用作分子篩、硅酸鹽耐火材料制品等的成型粘結劑及酒精脫水制乙烯和環氧乙烷的催化劑等[1-2]。擬薄水鋁石的工業生產方法主要有碳化法、鋁鹽中和法和醇鋁法[3],根據生產工藝不同,擬薄水鋁石中雜質含量變化很大。由于碳化法原料獲取簡單,生產成本低,目前國內主要采用向氧化鋁生產流程中的鋁酸鈉溶液中通入CO2制取擬薄水鋁石[3-5],此法的缺點之一是原料鋁酸鈉溶液成分復雜,雜質含量大[6-7],這些雜質元素會隨生產過程進入產品,大量有害雜質會使催化劑中毒,降低催化效率[3]。
目前,擬薄水鋁石化學分析方法還未有國家標準或行業標準規范,各企業主要參考YS/T 534.3~534.5《氫氧化鋁化學分析方法》測定SiO2、Fe2O3、Na2O含量,其中SiO2、Fe2O3含量采用分光光度法測定,Na2O含量采用火焰分光光度法或火焰原子吸收分光光度法測定。文獻[8-9]報道了用火焰原子吸收分光光度法測定擬薄水鋁石中的鐵,文獻[10]報道了用X熒光光譜法測定擬薄水鋁石中的鈣,而擬薄水鋁石中多種雜質的同時測定目前還未見報道。X射線熒光光譜法因具有分析速度快、可以同時測定多元素、準確度和精密度較高等特點而被廣泛應用于無機元素的檢測[11-13]。本研究采用與擬薄水鋁石成分相近的氫氧化鋁和氧化鋁國家標準物質繪制工作曲線,高頻熔樣,X熒光光譜法測定擬薄水鋁石中SiO2、Fe2O3、Na2O、K2O、CaO、ZnO、TiO2、V2O5、P2O5等微量和痕量雜質含量,并將測定結果與ICP-AES法和AAS法等的測定結果進行對比。
1 試驗部分
1.1 儀器與試劑
試驗儀器:Bruker S8 Tiger型波長色散X射線熒光光譜儀(德國布魯克公司),配有超尖銳端窗Rh陶瓷光管,75 μm鈹窗,真空光路,高壓發生器最大輸出功率4 kW,輸出電壓20~60 kV,輸出電流5~170 mA;Analymate-V4D型高頻熔樣機(北京靜遠世紀科技有限責任公司),額定功率4 kW,溫度范圍0~1 400℃;鉑-金坩堝(95%Pt+5%Au)。
無水四硼酸鋰-偏硼酸鋰混合熔劑(67%+33%,洛陽特耐實驗設備有限公司),溴化鋰溶液(300 g/L,國藥集團化學試劑有限公司),P10氣體(90%Ar+10%CH4)。
氫氧化鋁國家標準物質(GSB04-1814—2005~GSB04-1818—2005)5個(山東省冶金科學研究院),氧化鋁國家標準物質(Al2O3-01~Al2O3-10)6個(中國鋁業廣西分公司中心試驗室)。
四硼酸鋰-偏硼酸鋰混合熔劑為優級純,溴化鋰為分析純。
1.2 儀器測定條件
光譜儀為真空模式。定點測定時的樣品自轉速度為0.5 r/s,準直器面罩28 mm,譜峰和時間測定模式固定,無初級濾光片。各元素測定條件見表1。
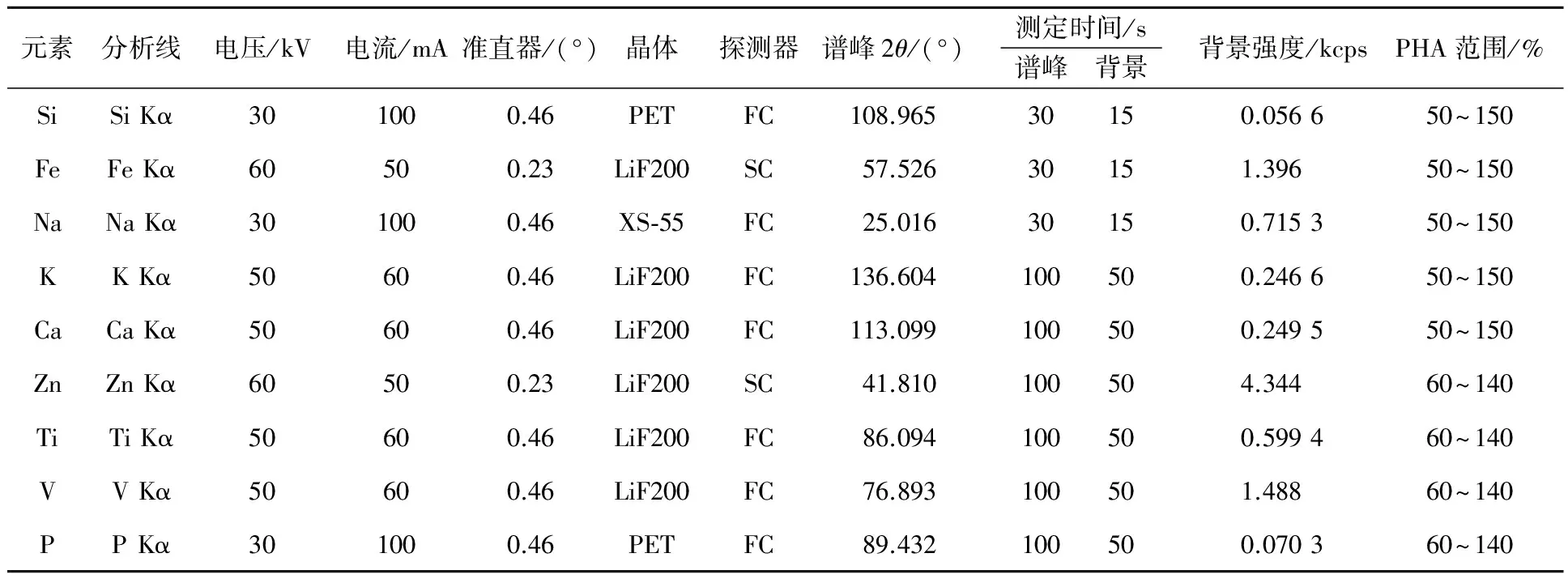
表1 各元素測定條件
注:FC為流氣式探測器(flow counter);SC為閃爍式探測器(scintillation counter)。
1.3 試驗方法
1.3.1 標準樣品
擬薄水鋁石目前還沒有國家標準物質,自制標準樣品工作量較大,且準確度難以保證。因此,選擇與擬薄水鋁石基體和雜質成分相近的氫氧化鋁和氧化鋁國家標準物質作為標準樣品,但由于這3種物質燒失量(灼減)各不相同,如果稱樣量相同,最終的熔片質量會存在較大差異,所以,根據各自的燒失量進行稱樣量校正,最終使熔片總質量保持一致。
1.3.2 燒失量測定
稱取在110 ℃下烘干的擬薄水鋁石樣品2.000 0 g于(1 100±20) ℃下灼燒30 min,以失去的質量計算燒失量。11個氫氧化鋁和氧化鋁國家標準物質已給出燒失量定值,無需測定。
1.3.3 玻璃片制備
稱取在110 ℃下烘干的氫氧化鋁標準樣品3.000 0 g,氧化鋁標準樣品和擬薄水鋁石樣品質量按式(1)計算稱取,再稱取混合熔劑6.000 0 g,一同置于鉑金坩堝中,混合均勻。然后滴加8滴溴化鋰溶液,于自動高頻熔樣機中在設定的熔樣條件下熔融14 min。熔樣過程中如發現有氣泡未趕凈或坩堝壁沾有試樣,可用坩堝鉗(夾持部分鉑金包裹)手動搖動。為減少二次污染,不使用鉑金模具。待熔融玻璃片冷卻后,在靠近坩堝的一面貼上標簽,光滑無裂紋的上表面作為分析面,保存于干燥器中,待測。
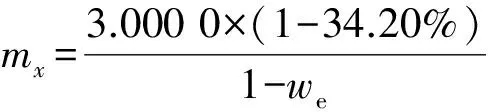
(1)
式中:mx為氧化鋁標準樣品或擬薄水鋁石樣品質量,g;3.000 0為氫氧化鋁標準樣品質量,g;34.20%為5個氫氧化鋁標準樣品燒失量平均值;we為氧化鋁標準樣品或擬薄水鋁石樣品的燒失量,%。
2 試驗結果與討論
2.1 稀釋比和樣品質量的確定
5個氫氧化鋁標準樣品燒失量為34.14%~34.26%,6個氧化鋁標準樣品燒失量為0.76%~0.98%,3個擬薄水鋁石樣品燒失量為22.52%~27.47%。5個氫氧化鋁標準樣品燒失量最大差值只有0.12%,所以保持氫氧化鋁標準樣品質量一致。分別按氫氧化鋁和混合熔劑稀釋比1∶3、1∶2、2∶3進行熔樣。結果表明:當稀釋比為2∶3時,氫氧化鋁在1 200 ℃下都很難完全熔融;當稀釋比為1∶2和1∶3時,在適當溫度和一定時間條件下可以熔融完全。由于氫氧化鋁、氧化鋁和擬薄水鋁石中雜質含量都很小,為了提高測定準確度,盡量減小混合熔劑與樣品的稀釋比,所以選擇稀釋比為1∶2,即氫氧化鋁質量3.000 0 g,混合熔劑質量6.000 0 g。氧化鋁標準樣品和擬薄水鋁石樣品質量按式(1)計算。
熔融結果表明:5個氫氧化鋁標準樣品玻璃片質量在7.941 8~7.957 8 g之間,6個氧化鋁標準樣品玻璃片質量在7.926 2~7.941 3 g之間,3個擬薄水鋁石樣品玻璃片質量在7.938 9~7.947 6 g之間,14個玻璃片質量最大值與最小值相對偏差小于0.4%,與理論計算值相符。
2.2 熔樣條件的確定
Analymate-V4D型自動高頻熔樣機采用分段加熱,各段溫度和時間、搖擺速度可以自行調節,設定好后自動運行。選擇自冷時間120 s,搖擺速度10(相對值,范圍為1~50),不同模式下的熔樣條件及結果見表2。
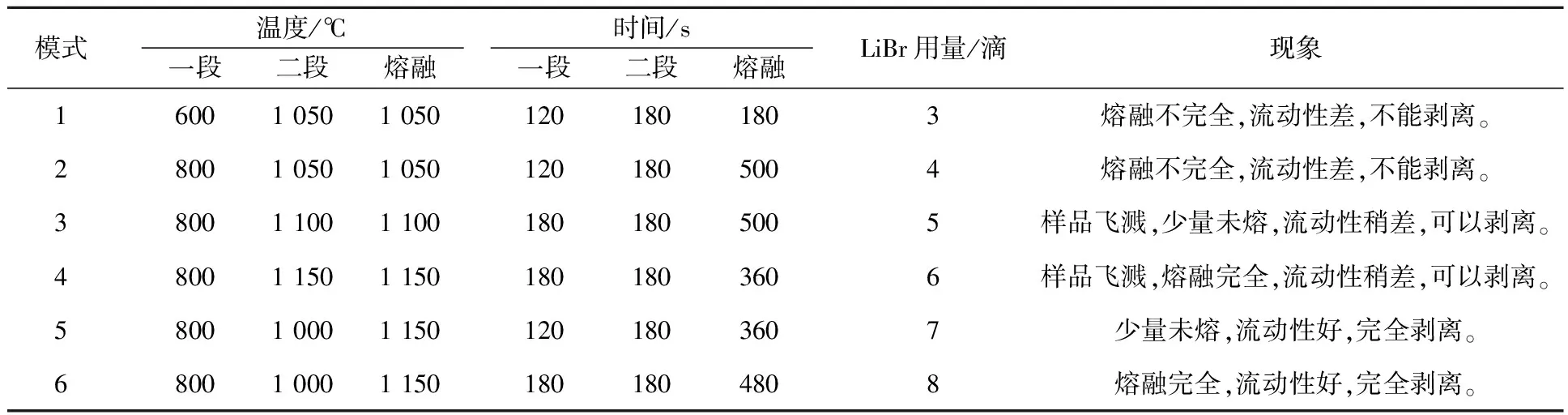
表2 不同模式下的熔樣條件及結果
從表2看出:當二段加熱溫度≥1 100 ℃時,未完全熔融樣品會飛濺;溴化鋰溶液用量≥7滴,熔融樣品流動性好,剝離容易。選擇模式6為最佳熔樣條件。
2.3 基體效應和譜線重疊校正
基體效應主要包含顆粒效應、礦物效應和元素間的吸收增強效應。采用熔融制樣可有效消除顆粒效應和礦物效應。元素間的吸收增強效應采用變化的理論α系數校正,校正公式見式(2),譜線重疊校正公式見式(3):
wi=s×(1+∑αij×wj)+b;
(2)
wi=s×(Ii+βij×Ik)×(1+∑αij×wj)+b。
(3)
式中:wi、wj分別為測定元素和影響元素的質量分數,%;s,b分別為校準曲線的斜率和截距;αij、βij分別為理論α系數和譜線重疊校正系數;Ii、Ik分別為測定元素的X熒光強度和重疊譜線的理論計算強度。
2.4 校準曲線和檢出限
按試驗方法,用11個氫氧化鋁和氧化鋁國家標準樣品制備玻璃片,測定玻璃片熒光強度,以凈強度(K)為縱坐標,質量分數為橫坐標繪制校準曲線。明顯偏離校準曲線的點不參與校準曲線的回歸。6個氧化鋁標準樣品未提供K2O和CaO標準值,這2種成分用5個氫氧化鋁標準樣片校正。由于燒失量和樣品質量不同,氧化鋁標準樣品錄入校準曲線的各成分標準值需按氫氧化鋁樣品質量換算(見式(4))。擬薄水鋁石各成分化學含量計算公式為:
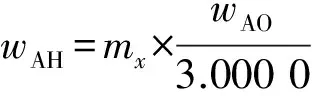
(4)
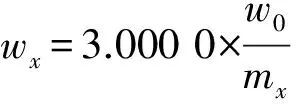
(5)式中:wAH為氧化鋁標準樣品各成分質量分數換算后的標準值,%;mx為氧化鋁標準樣品或擬薄水鋁石樣品質量,g;wAO為氧化鋁標準樣品各成分給定的標準質量分數,%;3.000 0為氫氧化鋁標準樣品質量,g;ws為擬薄水鋁石樣品各成分質量分數,%;w0為擬薄水鋁石各成分按校準曲線測定的質量分數,%。
校準曲線參數和檢出限見表3,檢出限計算公式為
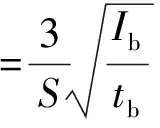
(6)
式中:S為測定靈敏度,即質量分數每變化1%引起的X射線熒光強度變化;Ib為背景X射線熒光強度;tb為背景測定時間,s。
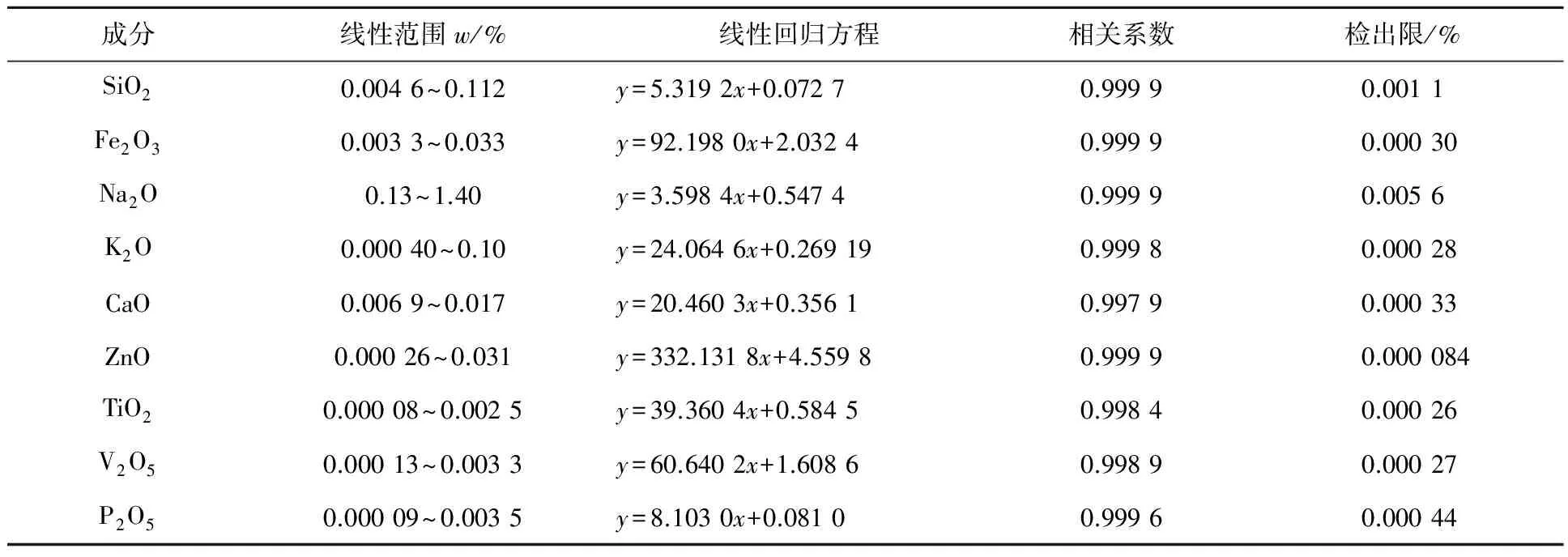
表3 校準曲線參數及檢出限
2.5 準確度和精密度
對2個不同的擬薄水鋁石樣品,在選定的試驗條件下測定各雜質質量分數,平行測定10次。擬薄水鋁石沒有標準樣品,所以其準確度采用與ICP-AES法和AAS法對比來確定,對比結果見表4。可以看出:當雜質質量分數大于0.001%時,本法測定值與ICP-AES法和AAS法測定值相吻合;當雜質質量分數小于0.001%時,本法與ICP-AES法和AAS法的測定值誤差小于GB/T 6609.30所要求的允許值;各成分10次測定的相對標準偏差在0.36%~8.3%之間。
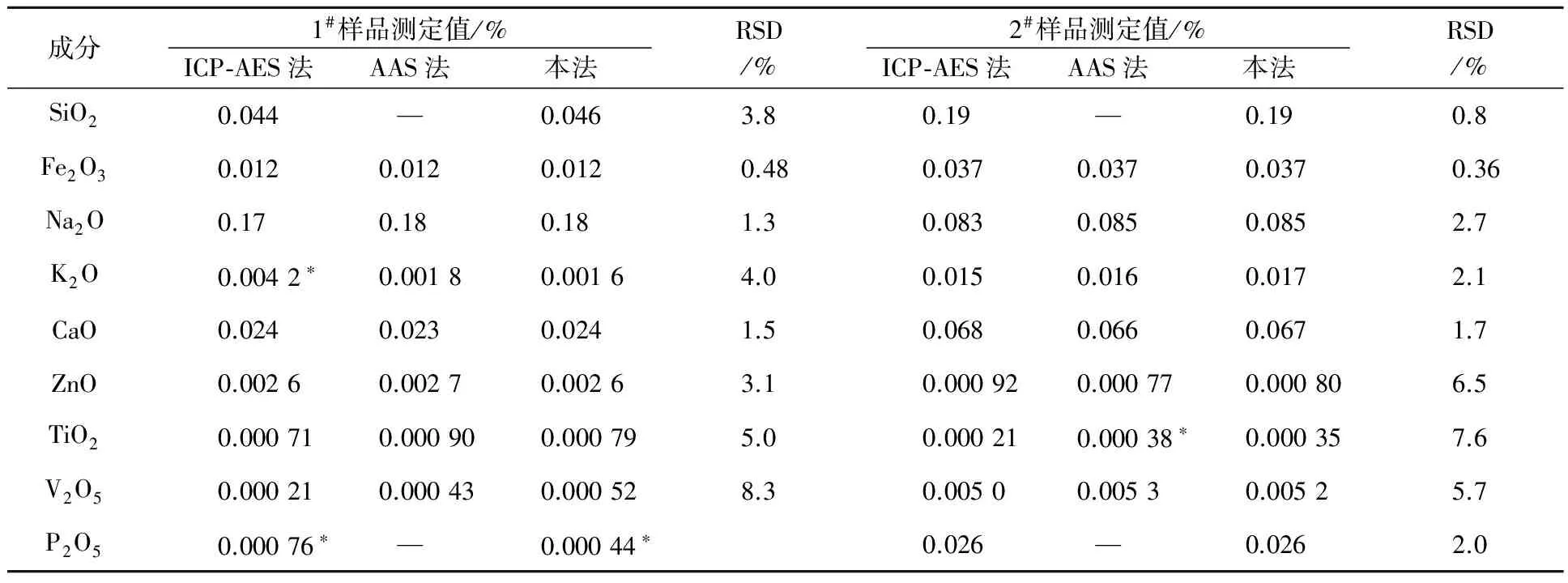
表4 測定方法的準確度和精密度(n=10)
注:*為該方法檢出限,%;—為未使用此方法。
3 結論
采用四硼酸鋰-偏硼酸鋰混合熔劑熔融樣品,有效消除礦物效應與顆粒效應的影響。由于氫氧化鋁和氧化鋁標準樣品與擬薄水鋁石樣品的燒失量不同,通過不同樣品質量校正了熔融玻璃片的質量差異。經過驗證,此方法準確度和精密度較高,實際樣品的測定值與ICP-AES法和AAS法測定結果相符,可以滿足對產品質量的分析需求。
[1] 苗壯,史建公,郝建薇,等.擬薄水鋁石的膠溶性與結構的關系[J].石油學報(石油加工),2016,32(3):493-500.
[2] 馬兵兵.X射線衍射法測定擬薄水鋁石的結晶度[J].理化檢驗(物理分冊),2015,51(7):474-476.
[3] 高建峰,徐春彥,王建中,等.用偏鋁酸鈉直接制取高純擬薄水鋁石[J].催化學報,2003,24(7):505-508.
[4] 王玉.鋁酸鈉溶液中和法制取擬薄水鋁石[J].輕金屬,2010(4):19-20.
[5] 曾豐,楊清河,曾雙親.采用NaAlO2-CO2連續中和法制備擬薄水鋁石[J].石油學報(石油加工),2015,31(5):1069-1074.
[6] 馬兵兵,蘇中華,彌海鵬,等.間接碘量法測定氧化鋁生產流程樣品鋁酸鈉溶液中硫離子、硫代硫酸根和亞硫酸根[J].冶金分析,2016,36(11):41-45.
[7] 馬兵兵.ICP-AES法測定鋁酸鈉溶液中的總硫和低價硫[J].濕法冶金,2017,36(1):79-82.
[8] 申明樂.火焰原子吸收光譜法測定擬薄水鋁石中鐵[J].光譜試驗室,2010,27(3):945-946.
[9] 賀曉東,黃安平.原子吸收法測定擬薄水鋁石中微量鐵[J].山西化工,2006,26(4):57-59.
[10] 潘志爽,劉明霞,王亞紅,等.擬薄水鋁石中雜質鈣含量的快速分析[J].石化技術與應用,2013,31(1):63-65,70.
[11] 王智鵬,李可及,董欣楊,等.六偏磷酸鈉熔融-X射線熒光光譜法測定鉻鐵礦主成分[J].濕法冶金,2016,35(1):83-86.
[12] 白萬里,張愛芬,石磊,等.X射線熒光光譜法測定工業硅中11種微量元素[J].冶金分析,2016,36(10):40-46.
[13] 包楚才,劉付建,劉瓊,等.波長色散X射線熒光光譜法快速測定內外墻涂料中鈦、鈣、鋇、鋅、鎂和硅[J].理化檢驗(化學分冊),2016,52(10):1178-1180.
Determination of Micro- and Trace-impurities in Pseudoboehmite by High-frequency Fusion and X-ray Fluorescence Spectrometer
MA Bingbing
(ChongqingAcademyofMetrologyandQualityInspection,Chongqing400020,China)
The high-frequency fusion and X-ray fluorescence spectrometer(XRFS) is applied to determine nine micro- and trace-impurities such as SiO2,Fe2O3,Na2O,K2O,CaO,ZnO,TiO2,V2O5,P2O5in pseudoboehmite.Using Li2B4O7and LiBO2as mixed fusion agent,LiBr as release agent,the sample is heated at 800 ℃ and 1 000 ℃ for 3 min,then fused at 1 150 ℃ for 8 min.After cooling,the glassy melt obtained is determined by XRFS.The calibration curve is plotted using alumina and aluminum hydroxide standard materials which are similar to pseudoboehmite in matrix and impurities,and the standard samples are fused into glassy melts in the same condition.Linear relationships between fluorescence intensities and chemical contents are kept in definite ranges.The determination values by the method are consistent with the determination values of ICP-AES method and AAS method when the impurities content is greater than 0.001%.The detection limits of the nine impurities components are found in the range of 0.000 084%-0.005 6%,and RSD(n=10) is in the range of 0.36%-8.3%.
high-frequency fusion;X-ray fluorescence spectrometer;pseudoboehmite;micro;trace;impurity
2016-12-21
馬兵兵(1986-),男,山西呂梁人,本科,工程師,主要研究方向為化學分析。E-mail:mabing1986310@qq.com。
O657.34
A
1009-2617(2017)04-0350-05
10.13355/j.cnki.sfyj.2017.04.022
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
產品可靠性報告(2017年7期)2017-09-05 09:49:12
專用汽車(2016年4期)2016-03-01 04:13:43
汽車觀察(2016年3期)2016-02-28 13:16:26
